一种单晶核壳结构高镍富钴正极材料的合成方法
1.本发明涉及锂离子电池领域,具体涉及一种单晶核壳结构高镍富钴正极材料的合成方法。
背景技术:
2.随着新能源时代的步伐越迈越快,世界各国的新能源行业迅猛发展。其中以储能为代表的锂离子电池成为了新能源行业发展的重中之重。尤其是汽车领域,以我国为例,像小鹏、蔚来、比亚迪等新能源汽车制造商蜂拥而至,他们将共同撑起中国新能源汽车发展的旗帜。电动汽车中的锂离子电池正极材料以磷酸铁锂与三元材料为主。
3.如今,高镍三元材料因其具有高超的能量密度以及功率密度将成为下一代高能量密度动力电池正极材料的首选。材料能量密度超过800wh/kg,8系高镍三元材料有望帮助动力电池系统能量密度超越200wh/kg。在三元正极材料中,人们往往忽略了co的重要性而一味地想要降低co含量盲目提升ni含量以达到能量密度/成本的最大化,从而导致8系高镍正极材料的循环稳定性下降。
技术实现要素:
4.本发明的目的是坚持以富钴的高镍三元材料出发,提出一个用核壳结构来应对富钴高镍三元材料不足的发明。
5.本发明的目的采用以下技术方案来实现:
6.一种单晶核壳结构高镍富钴正极材料的合成方法,包括以下步骤:
7.步骤(1)、制备核结构:
8.以聚乙二醇的水溶液作为溶剂、尿素作为沉淀剂,加入至以ni
0.82
co
0.12
mn
0.06
co3中金属元素的化学计量比配置的第一金属盐溶液中,然后转移到水热反应釜内反应,即得到核-ni
0.82
co
0.12
mn
0.06
co3;
9.步骤(2)、制备核壳结构的前驱体:
10.将核-ni
0.82
co
0.12
mn
0.06
co3加入至反应釜中作为共沉淀反应釜底液,然后按照壳-ni
0.75
co
0.15
mn
0.1
(oh)2中金属元素的化学计量比配置的第二金属盐溶液进料,经过共沉淀反应、陈化、抽滤、洗涤、干燥后,得到具有核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
。
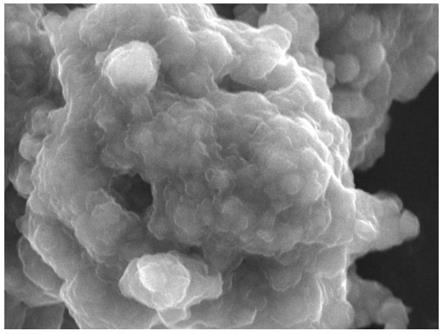
11.步骤(3)、将得到的具有核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
混锂后转移到马弗炉中,升温进行烧结处理,之后冷却到室温,得到单晶核壳结构的高镍富钴材料lini
0.8
co
0.13
mn
0.07
o2。
12.优选地,所述步骤(1)中,第一金属盐包括钴盐、镍盐和锰盐,第一金属盐溶液的浓度为0.2~0.5mol/l。
13.优选地,所述步骤(1)中,第一金属盐为硫酸盐、硝酸盐、氯化物、醋酸盐中的至少一种。
14.优选地,所述步骤(1)中,聚乙二醇为聚乙二醇200、聚乙二醇400、聚乙二醇600中
的至少一种。
15.优选地,所述步骤(1)中,聚乙二醇的水溶液中,聚乙二醇与水的体积比为1~5:1~3。
16.优选地,所述步骤(1)中,尿素用量是第一金属盐的总摩尔数的2~4倍。
17.优选地,所述步骤(1)中,水热反应釜的温度设置为180~200℃,反应时间为8~12h。
18.优选地,所述步骤(2)中,共沉淀反应过程中还加入了氢氧化钠作为沉淀剂、氨水作为络合剂,氢氧化钠的浓度为2mol/l,氨水的浓度为0.1mol/l,氢氧化钠的加入量是第二金属盐摩尔量的两倍,氨水的加入使反应体系ph调节为11.5
±
0.5。
19.优选地,所述步骤(2)中,第二金属盐溶液的浓度为1mol/l。
20.优选地,所述步骤(2)中,共沉淀反应的时间为12h,陈化时间为12h。
21.优选地,所述步骤(3)中,混锂使用的是氢氧化锂,混锂的过程中,具有核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
与氢氧化锂的摩尔比为1:1.05。
22.优选地,所述步骤(3)中,烧结处理的条件为:在氧气的氛围下,以5℃/min的速度升温至480℃,保温处理6h后,在750℃条件下退火18h。
23.本发明的有益效果为:
24.本发明利用一种巧妙的方法,对电池正极材料进行核壳处理,改变高镍层状材料的内部构成,抑制电池4.2v的非稳态相变,提升电池的循环性能。
25.本发明坚持以富钴的高镍三元材料出发,提出一个用核壳结构来应对富钴高镍三元材料不足的发明。本发明大大提升了8系高镍正极材料的循环稳定性,使其在200圈循环后仍然具有83%的保持率,相比之下lini
0.8
co
0.1
mn
0.1
o2(ncm811)的保持率不足50%。
26.本发明通过溶剂热-共沉淀复合方法合成一种单晶且具有核壳结构的高镍富钴层状正极材料。本发明能有效抑制高镍层状材料在4.2v处的非稳态相变,且抑制了微裂纹由颗粒内向表面蔓延,大大提升了其循环性能。该策略优点在于从根本上解决了高镍三元材料循环衰减严重的问题,效果显著,对规模化工业生产的意义重大。
附图说明
27.利用附图对本发明作进一步说明,但附图中的实施例不构成对本发明的任何限制,对于本领域的普通技术人员,在不付出创造性劳动的前提下,还可以根据以下附图获得其它的附图。
28.图1为实施例1制备的lini
0.8
co
0.13
mn
0.07
o2材料前驱体的扫描电镜图。
29.图2为实施例1制备的lini
0.8
co
0.13
mn
0.07
o2材料的扫描电镜图。
30.图3为对比实施例制备的lini
0.82
co
0.12
mn
0.06
o2材料前驱体扫描电镜图。
31.图4为对比实施例制备的lini
0.82
co
0.12
mn
0.06
o2材料扫描电镜图。
32.图5为实施例1制备的1lini
0.8
co
0.13
mn
0.07
o2材料全局电子衍射。
33.图6为实施例1、对比实施例与原始ncm811的循环性能图。
34.图7为实施例1、对比实施例与原始ncm811的倍率性能图。
35.图8为实施例1、对比实施例与原始ncm811的经典交流阻抗谱。
36.图9为实施例1、对比实施例、原始ncm811和标准卡pdf#85-1978#的xrd图谱。
37.图10为实施例1制备的lini
0.8
co
0.13
mn
0.07
o2材料循环500圈后的横截面扫描电镜图。
38.图11为原始ncm811材料循环500圈后的横截面扫描电镜图。
具体实施方式
39.为了更清楚的说明本发明,对本发明的技术特征、目的和有益效果有更加清楚的理解,现对本发明的技术方案进行以下详细说明,但不能理解为对本发明的可实施范围的限定。
40.本发明公开了一种改变锂离子电池正极材料表面结构的策略,为展示本发明对改变锂离子电池表面结构的效果,设立以下实施例和对照例。
41.实施例1
42.本实施例是单晶核壳结构材料的制备及扣式电池测试,包括以下步骤:
43.1)利用聚乙二醇辅助溶剂热法合成核-ni
0.82
co
0.12
mn
0.06
co3(前驱体,球型):聚乙二醇与水调配成体积比为2:1的溶液作为溶剂,尿素以两倍的金属盐摩尔量加入以化学计量比配好金属盐溶液中搅拌使其溶解,然后转移到水热反应釜,在200℃下反应12h,经过抽滤、纯水洗涤三次,80℃真空干燥,得到ni
0.82
co
0.12
mn
0.06
co3(前驱体);
44.2)将核-ni
0.82
co
0.12
mn
0.06
co3加入至反应釜中作为共沉淀反应釜底液,进料时,金属盐按照壳-ni
0.75
co
0.15
mn
0.1
(oh)2中金属元素的化学计量比进料,控制核:壳的摩尔比为7:3,共沉淀反应12h、陈化12h、用纯水洗涤三遍、80℃真空干燥后,得到具有核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
(lini
0.8
co
0.13
mn
0.07
o2材料前驱体);
45.3)将得到核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
混锂,核壳结构的前驱体与氢氧化锂的混合比为1:1.05,之后转移到马弗炉,以5℃/min的速度升温到480℃预烧6h,然后升至750℃退火18h,最终冷却到室温,得到单晶核壳结构的高镍富钴材料lini
0.8
co
0.13
mn
0.07
o2;
46.4)将步骤3)所得到的lini
0.8
co
0.13
mn
0.07
o2材料、导电石墨super p与聚偏氟乙烯pvdf以质量比8:1:1的混合并分散到氮甲基吡咯烷酮中,调节固含量为45%-50%,以1000rpm的转速搅拌30min,得到均匀的正极材料浆料,再用100微米涂布器将上述浆料均匀涂布在14微米厚的铝箔上,然后用冲片机裁出若干12mm直径的圆片,即得到正极片,并组装成以锂片为负极的扣式电池cr2032中进行电化学性能测试。
47.附图1,附图2分别为该实施例所得的lini
0.8
co
0.13
mn
0.07
o2材料前驱体(ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
)以及本体的扫描电镜图,可以看到烧结后材料表面模糊,有明显的附着物。
48.实施例2
49.本实施例是单晶核壳结构材料的制备及扣式电池测试,包括以下步骤:
50.1)利用聚乙二醇辅助溶剂热法合成核-ni
0.85
co
0.11
mn
0.04
co3(前驱体,球型):聚乙二醇与水调配成体积比为1:1的溶液作为溶剂,尿素以1.5倍的金属盐摩尔量加入以化学计量比配好金属盐溶液中搅拌使其溶解,然后转移到水热反应釜,在180℃下反应15h,抽滤,纯水洗涤三次,80℃真空干燥得到ni
0.85
co
0.11
mn
0.04
co3;
51.2)将核-ni
0.85
co
0.11
mn
0.04
co3(前驱体)加入至反应釜中作为共沉淀反应釜底液,进
料时,金属盐按照壳-ni
0.75
co
0.15
mn
0.1
(oh)2中金属元素的化学计量比进料,控制核:壳的摩尔比为6:4,共沉淀反应12h、陈化12h、用纯水洗涤三遍、80℃真空干燥后,得到具有核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
(lini
0.8
co
0.13
mn
0.07
o2材料前驱体);
52.3)将得到核壳结构的前驱体ni
0.8
co
0.13
mn
0.07
(co3)
x
((oh)2)
1-x
混锂,核壳结构的前驱体与氢氧化锂的混合比为1:1.05,之后转移到马弗炉,以5℃/min的速度升温到480℃预烧6h,然后升至750℃退火18h,最终冷却到室温,得到单晶核壳结构的高镍富钴材料lini
0.8
co
0.13
mn
0.07
o2;
53.4)将步骤3)所得到的lini
0.8
co
0.13
mn
0.07
o2材料与导电石墨super p与聚偏氟乙烯pvdf以质量比8:1:1的混合并分散到氮甲基吡咯烷酮中,以1000rpm的转速搅拌30min,得到均匀的正极材料浆料,再用100微米涂布器将上述浆料均匀涂布在14微米厚的铝箔上,然后用冲片机裁出若干12mm直径的圆片,即得到正极片,并组装成以锂片为负极的扣式电池cr2032中进行电化学性能测试。
54.对比实施例
55.本对比实施例是单晶核壳结构的核部分材料lini
0.82
co
0.12
mn
0.06
o2制备及扣式电池测试,包括以下步骤:
56.1)利用聚乙二醇辅助溶剂热法合成核-ni
0.82
co
0.12
mn
0.06
co3(前驱体,球型),聚乙二醇与水调配成体积比为2:1的溶液作为溶剂,尿素以2倍的理论用量加入以化学计量比配好金属盐溶液中搅拌使其溶解,然后转移到水热反应釜,在200℃下反应12h,抽滤,纯水洗涤三次,80℃真空干燥得到核结构前驱体ni
0.82
co
0.12
mn
0.06
co3;
57.2)将得到核结构的ni
0.82
co
0.12
mn
0.06
co3混锂,核结构与氢氧化锂的混合比为1:1.05,之后转移到马弗炉,以5℃/min的速度升温到480℃预烧6h,然后升至750℃退火18h,最终冷却到室温,得到单晶富钴高镍材料lini
0.82
co
0.12
mn
0.06
o2(材料lini
0.82
co
0.12
mn
0.06
o2前驱体);
58.3)将步骤2)所得到的lini
0.82
co
0.12
mn
0.06
o2材料与导电石墨super p与聚偏氟乙烯pvdf以质量比8:1:1的混合并分散到氮甲基吡咯烷酮中,以1000rpm的转速搅拌30min,得到均匀的正极材料浆料,再用100微米涂布器将上述浆料均匀涂布在14微米厚的铝箔上,然后用冲片机裁出若干12mm直径的圆片,即得到正极片,并组装成以锂片为负极的扣式电池cr2032中进行电化学性能测试。
59.附图3,附图4分别为该对比实施例所得的lini
0.82
co
0.12
mn
0.06
o2材料前驱体以及本体的扫描电镜图,可以看到烧结后材料表面棱角分明,轮廓清晰与实施例1形成了鲜明的对比。
60.此外,原始ncm811与商用的ncm811也一起与本发明的实施例进行对比。
61.为了更加清楚的说明本发明,本发明还做了图5~图11的对比实验,具体如下:
62.图5为实施例1的核壳结构的富钴高镍层状材料lini
0.8
co
0.13
mn
0.07
o2的全局电子衍射,从图中清晰的光斑可以证明所合成的材料为单晶。
63.图6为不同样品的循环性能图,实施例1的样品在循环200圈后依然保持83%的保持率,此外没有核壳的单纯富钴高镍材料的循环保持率为70%。然而商用的ncm811以及原始的ncm811在经过200圈循环后保持率分别只有55%以及45%。
64.从图7的倍率性能图中看出,在20c的高倍率下,实施例1的单晶核壳材料依然能放
出90以上的克容量。相比之下原始ncm811在此倍率下容量保持率接近0。
65.图8的交流阻抗普也可以看出,实施例1所制备的单晶核壳材料具有最小的电荷转移阻抗。
66.图9的各组材料xrd图表示,没有新的特征峰的出现,表明材料在原ncm811的基础上相差不大。
67.图10为实施例1、2所制备的单晶核壳富钴高镍材料循环500圈后的横截面sem图,可以看到核的崩坏并没有延伸到颗粒表面,颗粒的形态学得到了维持;
68.图11中能够看出,原始ncm811的横截面裂纹已经蔓延到颗粒的表面了,事实上颗粒的内部结构已经坍塌。
69.最后应当说明的是,以上实施例仅用以说明本发明的技术方案,而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细地说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1