改性二氧化硅、其制备方法及其用途与流程
改性二氧化硅、其制备方法及其用途
1.本发明涉及改性二氧化硅、其制备方法及其用途。
2.e.f.vansant,p.van der voort,k.c.vrancken,characterization and chemical modification of the silica surface,chemical studies in surface science and catalysis vol.93,elsevier verlag,1995公开了使用各种试剂例如有机硅烷对二氧化硅的化学改性(第194-198页;第266-270页;第288-292页)。
3.从de 10138491 a1和de 10138492 a1中也可知沉淀二氧化硅的疏水化,其中直接加入到干燥器。另外还有一个调理步骤。
4.wo2014033300 a1公开了使用二羧酸对沉淀二氧化硅进行改性。
5.已知改性二氧化硅的缺点是混合物的粘度升高,从而橡胶混合物的加工较差。此外,这些还伴随着加工窗口缩小和过早交联的趋势,这使制备中的加工更加复杂。预处理导致的另一个缺点是对动态特性的不利影响,以及硫化材料中填料的分散变差。
6.本发明的目的是提供一种改性二氧化硅,通过与原位硅烷化或原位改性进行比较,所述改性二氧化硅显示混合物加工特性的改善以及动态特性的改善。此外,改性二氧化硅具有等同或改善的填料分散的质量。
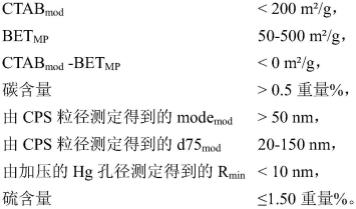
7.本发明提供了改性二氧化硅,其特征在于具有以下的理化参数:
[0008][0009]
根据本发明的改性二氧化硅》150μm下的ro-tap值可以是80%,优选》85%。
[0010]
根据本发明的改性二氧化硅》300μm下的ro-tap值可以是50%,优选》60%。
[0011]
根据本发明的改性二氧化硅》500μm下的ro-tap值可以是20%,优选》40%。
[0012]
根据本发明的改性二氧化硅的doa吸收量(doaabsorption)可以是100-300ml/(100g),优选140-240ml/(100g)。
[0013]
根据本发明的改性二氧化硅的干燥损失可以是《4.5重量%,优选2.0-4.0重量%。
[0014]
根据本发明的改性二氧化硅的ph值可以是≥6.3,优选6.3-8.0。
[0015]
根据本发明的改性二氧化硅的tar
mod
值可以是》1%,优选15-70%。
[0016]
根据本发明的改性二氧化硅的灼烧残渣可以是70-95%,优选80-95%。
[0017]
由加压的hg孔径测定(hg pore size determination,pressurized)得到的根据本发明的改性二氧化硅的if值可以是优选
[0018]
由加压的hg孔径测定得到的根据本发明的改性二氧化硅的is值可以是≤79ml/(100g),优选50-79ml/(100g)。
[0019]
根据本发明的改性二氧化硅的pv值(v80,3.7-80nm,140
°
)可以是≤0.86ml/g,优选0.30-0.86ml/g。
[0020]
根据本发明的改性二氧化硅可以是气相二氧化硅或沉淀二氧化硅,优选沉淀二氧化硅。
[0021]
本发明还提供了一种制备根据本发明的改性二氧化硅的方法,其特征在于在干燥单元的入口将二氧化硅与至少一种选自以下的添加剂混合:水性含硫烷氧基硅烷乳液、聚硅氧烷如优选聚二甲基硅氧烷及其侧链改性衍生物、含硫烷氧基硅烷和聚二甲基硅氧烷的混合物,以及含硫烷氧基硅烷和阴离子聚醚的混合物,然后供给至干燥单元。
[0022]
已经在入口进行初步混合,这与de 10138491 a1和de 10138492 a1不同,在de 10138491 a1和de 10138492 a1中,将添加剂直接添加至干燥器中。此外,在本发明中,不需要申请de 10138491 a1和de 10138492a1中所述的后续调理步骤。
[0023]
所述改性二氧化硅无需进一步调理即可使用。
[0024]
入口可以是输送螺杆,优选双螺杆,更优选非啮合双螺杆(non-meshing twin screw)。
[0025]
根据本发明的方法中使用的二氧化硅的bet
mp
表面积可以是50-300m2/g,优选80-280m2/g。二氧化硅的ctab表面积可以是40-280m2/g,优选70-260m2/g。
[0026]
二氧化硅可以是气相二氧化硅或沉淀二氧化硅,优选沉淀二氧化硅。
[0027]
含硫烷氧基硅烷可以是式(i)的烷氧基硅烷
[0028]
[(ro)3si-r
3-]na (i)
[0029]
其中r相同或不同,并且是直链未取代或支链未取代的(c1-c10)烷基,优选(c1-c6)烷基,更优选甲基或乙基,或烷基聚醚基-(r
1-o)
m-r2,其中r1相同或不同,并且是支链或非支链的饱和脂族二价c1-c30烃基,m平均为1至30,r2是未取代的支链或非支链的单价c1-c30烷基、c2-c30烯基、c6-c30芳基或c7-c30芳烷基,r3是支链或非支链、饱和或不饱和、脂族、芳族或混合的脂族/芳族二价c1-c30烃基,n=1或2,当n=2时,a是s
x
,x=1-10,且当n=1时,a是sh、scn或sc(o)r。
[0030]
所述含硫烷氧基硅烷可以优选是双[(3-三乙氧基甲硅烷基)丙基]二硫化物或(eto)3si-(ch2)
3-s-c(o)-c7h
15
。
[0031]
所述水性含硫烷氧基硅烷乳液可以是式(i)的烷氧基硅烷的乳液。
[0032]
所述水性含硫烷氧基硅烷乳液可以优选是双[(3-三乙氧基甲硅烷基)丙基]二硫化物的乳液。使用的水性含硫烷氧基硅烷乳液可以是50重量%的双[(3-三乙氧基甲硅烷基)丙基]二硫化物或双[(3-三乙氧基甲硅烷基)丙基]多硫化物乳液,其具有《0.5重量%的非离子表面活性剂。
[0033]
所述聚硅氧烷可以是改性或非改性聚二甲基硅氧烷。改性聚二甲基硅氧烷可以含有聚醚磷酸酯基团、烷基酯基团或聚醚基团。使用的聚硅氧烷可以是聚醚硅氧烷。
[0034]
所述含硫烷氧基硅烷和聚二甲基硅氧烷的混合物可以含有0.1-50重量%的聚二甲基硅氧烷。
[0035]
所述含硫烷氧基硅烷和阴离子聚醚的混合物可以含有0.1-50重量%的聚醚。
[0036]
所述聚醚可以是阴离子改性聚醚;例如,所述聚醚可以含有磷酸酯基团。
[0037]
干燥单元可以是henschel混合器或旋转闪蒸干燥器。
[0038]
henschel混合器中的反应可以在80℃-170℃,优选95℃-145℃的温度下进行。
[0039]
旋转闪蒸干燥器中的反应可以在70℃-180℃,优选80℃-170℃的出口温度下进行。
[0040]
添加剂的初步分布不是像de 10138492 a1中所述的同步干燥,而是在干燥器上游的入口中进行。例如,这可以以高剪切、啮合双螺杆(meshing twin screw)的形式来完成。
[0041]
二氧化硅/添加剂重量比可以是100:1-100:15,优选100:3-100:10,更优选100:4-100:8。
[0042]
本发明还提供了一种橡胶混合物,其包含
[0043]
(a)橡胶或橡胶混合物,和
[0044]
(b)至少一种根据本发明的改性二氧化硅。
[0045]
所使用的橡胶可以是天然橡胶和/或合成橡胶。优选的合成橡胶例如描述在w.hofmann,kautschuktechnologie[rubber technology],genter verlag,stuttgart 1980中。它们可以包括:
[0046]-聚丁二烯(br),
[0047]-聚异戊二烯(ir),
[0048]-苯乙烯/丁二烯共聚物,例如乳液sbr(e-sbr)或溶液sbr(s-sbr),优选苯乙烯含量为1重量%-60重量%,更优选5重量%-50重量%(sbr),
[0049]-氯丁二烯(cr),
[0050]-异丁烯/异戊二烯共聚物(iir),
[0051]-丁二烯/丙烯腈共聚物,其丙烯腈含量为(nbr)的5重量%-60重量%,优选10重量%-50重量%,
[0052]-部分氢化或完全氢化的nbr橡胶(hnbr),
[0053]-乙烯/丙烯/二烯共聚物(epdm),
[0054]-上述橡胶,其还具有官能团,如羧基、硅醇或环氧基,例如环氧化nr、羧基官能化nbr或硅醇(-sioh)-或甲硅烷氧基(-si-or)官能化sbr,
[0055]
以及这些橡胶的混合物。
[0056]
在优选的实施方案中,橡胶是可以用硫来硫化的。对于汽车轮胎胎面的制造,特别可以使用玻璃化转变温度高于-50℃的阴离子聚合s-sbr橡胶(溶液sbr),以及这些与二烯橡胶的混合物。可以特别优选使用s-sbr橡胶,其中丁二烯组分的乙烯基含量超过20重量%。特别优选使用s-sbr橡胶,其中丁二烯组分的乙烯基含量超过50重量%。
[0057]
可以优选使用上述橡胶的混合物,其s-sbr含量超过50重量%,优选超过60重量%。
[0058]
根据本发明的橡胶混合物可以包含其他填料。以下填料可以用作根据本发明的橡胶混合物的填料:
[0059]-炭黑:炭黑可以通过灯黑法、炉黑法、气黑法或热处理法制备,并且bet表面积为20至200m2/g。炭黑也可以任选地含有杂原子,例如,如si。
[0060]-无定形二氧化硅,例如,通过从硅酸盐溶液中沉淀或通过卤化硅的火焰水解制
备,其比表面积为5至1000m2/g,优选20至400m2/g(bet表面积),初级粒径为5至400nm。二氧化硅也可以任选地是与其他金属氧化物的混合氧化物的形式,所述其他金属氧化物如al氧化物、mg氧化物、ca氧化物、ba氧化物、zn氧化物和钛氧化物,和/或含有痕量的至多10 000ppm的这些金属离子。
[0061]-合成硅酸盐(如硅酸铝)、碱土金属硅酸盐(如硅酸镁或硅酸钙),其bet表面积为20至400m2/g,初级颗粒直径为10至400nm。
[0062]-合成或天然铝氧化物和合成或天然氢氧化铝。
[0063]-天然硅酸盐,如高岭土和其他天然存在的二氧化硅。
[0064]-玻璃纤维和玻璃纤维产品(垫、线)或玻璃微珠。
[0065]
可以优选使用通过硅酸盐溶液的沉淀制备的无定形二氧化硅,其bet表面积为20至400m2/g,更优选100m2/g至250m2/g,其量为5至150重量份,每种情况下基于100份的橡胶。
[0066]
所述填料可以单独使用或以混合物的形式使用。
[0067]
所述橡胶混合物可以包含5至150重量份的根据本发明的改性二氧化硅,以及0.1至20重量份、优选1至18重量份、更优选5至15重量份的有机硅烷,其中重量份为基于100重量份的橡胶。
[0068]
所述橡胶混合物还可以包含硅油和/或烷基硅烷。所述橡胶混合物还可以包含树脂、改性树脂和/或反应性树脂。
[0069]
根据本发明的橡胶混合物可以包含其他已知的橡胶助剂,例如交联剂、硫化促进剂、反应促进剂、反应阻滞剂(reaction retarders)、抗氧化剂、稳定剂(包括老化稳定剂)、加工助剂、增塑剂、蜡或金属氧化物,以及任选存在的活化剂,如三乙醇胺、聚乙二醇或己三醇。
[0070]
根据包括最终用途在内的因素,橡胶助剂可按常规用量使用。常规用量例如可以是基于橡胶的0.1重量%至50重量%的量。
[0071]
使用的交联剂可以是硫或有机硫供体。
[0072]
根据本发明的橡胶混合物可以包含其他硫化促进剂。可以使用的合适的硫化促进剂的实例包括巯基苯并噻唑类、亚磺酰胺类、胍类、二硫代氨基甲酸盐类、硫脲类、硫代碳酸盐类以及这些的锌盐,例如二丁基二硫代氨基甲酸锌。
[0073]
根据本发明的橡胶混合物还可以包含:
[0074]
硫化秋兰姆(thiuram sulfide)促进剂和/或氨基甲酸酯促进剂和/或相应的锌盐,
[0075]
含氮共活化剂,
[0076]
任选存在的其他橡胶助剂,和
[0077]
任选存在的其他促进剂。
[0078]
促进剂与含氮共活化剂的按重量计的重量比可以等于或大于1。
[0079]
根据本发明的橡胶混合物可以包含基于100重量份的橡胶至少0.25重量份的四苄基秋兰姆二硫化物或四甲基秋兰姆二硫化物,基于100重量份的橡胶至多0.25重量份的二苯基胍、环己基亚磺酰胺或二环己基亚磺酰胺。
[0080]
可以优选地一起使用亚磺酰胺与胍和秋兰姆,更优选一起使用环己基亚磺酰胺或二环己基亚磺酰胺与二苯基胍和四苄基秋兰姆二硫化物或四甲基秋兰姆二硫化物。
[0081]
基于所使用的橡胶,硫化促进剂和硫的用量可以是0.1重量%至10重量%,优选0.1重量%至5重量%。可以特别优选使用硫和亚磺酰胺类,其量为1重量%至4重量%,秋兰姆类的量为0.2重量%至1重量%,胍类的量为0重量%至0.5重量%。
[0082]
本发明还提供了一种制备根据本发明的橡胶混合物的方法,其特征在于在混合单元中混合橡胶或橡胶混合物、根据本发明的改性二氧化硅以及任选存在的其他橡胶助剂。
[0083]
橡胶与填料、任何橡胶助剂和根据本发明的改性二氧化硅的共混可以在常用的混合单元中进行,如滚筒、密闭式混合机和混合挤出机。这种类型的橡胶混合物通常可以在密闭式混合机中制备,方法是通过首先在一个或多个连续的热机械混合阶段中在100℃至170℃下混合橡胶、填料、根据本发明的改性二氧化硅和橡胶助剂。各个组分的加入顺序和加入时机可能对混合物的最终性能产生关键影响。通常可以在40℃至110℃下,将所得橡胶混合物与交联化学品在密闭式混合机或辊上混合,并对混合物进行处理,以获得用于方法的后续步骤例如成型和硫化的所谓粗混合物。
[0084]
橡胶混合物可以以母料形式和/或连续混合工艺使用。橡胶混合物可以通过被称为液相混合或连续液相混合的途径,或通过母料的组合进行制备。
[0085]
根据本发明的橡胶混合物可以在80℃至200℃,优选130℃至180℃的温度下硫化,任选地在10至200bar的压力下硫化。
[0086]
根据本发明的橡胶混合物可以用于通过硫化制备成形体。
[0087]
本发明的橡胶混合物可以用于制备模具,例如用于制备充气轮胎、电缆护套、软管、传动带、输送带、辊覆盖物、轮胎(特别是轮胎胎面)、鞋底、密封元件,如密封圈和阻尼元件等。
[0088]
根据本发明的改性二氧化硅的优点是相应的橡胶混合物具有减小的门尼粘度、延长的加工窗口,从而提高了可加工性。此外,根据本发明的橡胶混合物显示出改善的动态性能和改善的断裂伸长率,以及硫化产物的相等的抗拉强度、分散质量和耐磨性。
[0089]
测试方法:
[0090]
ctab-根据iso 5794-1-g测定ctab表面积
[0091]
该方法基于水溶液中的缓冲ctab(n-十六烷基-n,n,n-三甲基溴化铵)在二氧化硅“外”表面上的吸附,所述“外”表面也称为“橡胶活性表面”。利用ndss(磺基琥珀酸二辛酯钠溶液),对未吸附的ctab进行反滴定。滴定终点为溶液不透明度的最大上升处。
[0092]
该程序符合iso 5794-1g,其说明、增补和偏离如下所述:
[0093]
·
在样品制备过程中,优选使用合适的研磨机对粗颗粒形式的二氧化硅和硅酸盐样本进行超细研磨,或使用研钵和研杵进行粉碎,并通过90μm筛进行筛分,而不是使用研钵和研杵进行粉碎并使用标准中描述的150μm筛进行分级。
[0094]
·
将由测试样本和预期ctab表面积小于200m2/g的ctab溶液组成的悬浮液搅拌10分钟。如标准中所述,将由测试样本和预期ctab表面积不小于200m2/g的ctab溶液组成的悬浮液搅拌35分钟。
[0095]
·
吸附后,通过0.2μm聚酰胺过滤器过滤二氧化硅。
[0096]
·
使用具有自动进样器和tirando 809的metrohm滴定处理器滴定滤液。使用的光度电极是metrohm的spectrosense 523nm。
[0097]
ctab
mod-根据iso 5794-1-g测定乙醇ctab值
–
ctab
mod.
[0098]
该方法基于水性乙醇溶液中的缓冲ctab(n-十六烷基-n,n,n-三甲基溴化铵)在疏水二氧化硅“外”表面上的吸附,所述“外”表面也称为“橡胶活性表面”。利用ndss(磺基琥珀酸二辛酯钠溶液),对未吸附的ctab进行反滴定。滴定终点为溶液不透明度的最大上升处。
[0099]
该程序符合iso 5794-1g,其说明、增补和偏离如下所述:
[0100]
·
在样品制备过程中,优选使用合适的研磨机(例如,ika m20研磨机)对粗颗粒形式的二氧化硅和硅酸盐样本进行超细研磨,或使用研钵和研杵进行粉碎,并通过75μm筛进行筛分,而不是使用研钵和研杵进行粉碎并使用标准中描述的150μm筛进行分级。
[0101]
·
为测定所用ndss溶液的空白值,移取(30.0
±
0.1)ml的ctab溶液和(10.0
±
0.1)ml的乙醇、以及55ml的软化水的混合物中的5.00ml,置于滴定烧杯中。
[0102]
·
称取分级的样品材料,精确到0.1mg,置于50ml具有磁力搅拌棒的离心管中,并用10.0ml乙醇预先润湿。随后加入(30.0
±
0.1)ml的ctab溶液,用磁力搅拌器搅拌20-25分钟(对于预期ctab值大于200m2/g的样品,则为35-40分钟)。
[0103]
·
通过在至少4000rpm和rcf 2500下离心15分钟,吸附后分离固相和液相。
[0104]
·
使用具有自动进样器和tirando 905的metrohm滴定处理器滴定液相。使用的光度电极是mettler-toledo的dp5,工作波长为555nm。
[0105]
bet
mp-根据din iso 9277测定氮bet表面积
[0106]
根据din iso 9277,该方法用于通过bet方法测定二氧化硅的n2比表面积。在该方法中,通过在规定分压下低温吸附氮气来进行该测量。该分析是作为多点测定进行的,在总共5个测量点测定的情况下,在分压范围(p/po)为0.05-0.2的情况下几乎显示出线性行为。
[0107]
该程序符合din iso 9277及下述说明:
[0108]
称取前,使用刮刀小心粉碎颗粒样品,然后使用micromeritics vac prep061真空脱气恒温器在(160
±
2)℃下减压脱气60分钟。
[0109]
为测定bet表面积,在吸附阶段期间记录以下5个相对压力点(p/po):0.0500;0.0875;0.1250;0.1625和0.2000。
[0110]
为了测量,使用来自micromeritics的tristar 3000系列(3000/3020/3030),使用静态体积试验方法以及杜瓦容器。
[0111]
碳含量
–
根据din en iso 3262-19测定碳含量
[0112]
根据din en iso 3262-19,使用leco的元素分析仪(仪器类型cs-200和cs-600)和外部评价单元(具有leco软件的pc)进行测定。
[0113]
称取样品,置于陶瓷坩埚中,加入氧化催化剂和燃烧促进剂(例如leco的lecocel ii)以及诱导剂(例如leco的超纯铁屑(ultrapure iron turnings)),在元素分析仪的感应烘箱中燃烧样品。样品中的碳在氧气流中氧化成co2。通过红外检测对气体进行定量。对于分析仪的操作、维护和调整,适用leco的操作手册中的指南和说明。
[0114]
使用bam钢标准号289-1、标准号130-1和标准号283-1完成校准。应选择标准品和起始重量,从而测试样本的硫质量在校准范围内。通过计算包含至少4个校准点的校准线,完成校准。
[0115]
硫含量-总硫-根据din en iso 3262-19或astm d 6741方法b测定二氧化硅测试样本
[0116]
a.未改性二氧化硅的测定方法
[0117]
根据din en iso 3262-19,使用leco的元素分析仪(仪器类型cs-200和cs-600)和外部评价单元(具有leco软件的pc)进行测定。
[0118]
称取样品,置于陶瓷坩埚中,加入氧化催化剂和燃烧促进剂(例如leco的lecocel ii)以及诱导剂(例如leco的超纯铁屑),在元素分析仪的感应烘箱中燃烧样品。样品中的硫在氧气流中氧化成so2。通过红外检测对气体进行定量。对于分析仪的操作、维护和调整,适用leco的操作手册中的指南和说明。
[0119]
使用bam钢标准号289-1、标准号130-1和标准号283-1完成校准。应选择标准品和起始重量,从而测试样本的硫质量在校准范围内。通过计算包含至少4个校准点的校准线,完成校准。
[0120]
b.改性二氧化硅的测定方法
[0121]
使用lecosc-144dr总硫分析仪测定硫含量。样品在氧气流中于1350℃下燃烧;通过红外测量单元对形成的so2进行定量。该方法符合astm d 6741方法b。
[0122]
测定时,称取测试样本和校准物质装入蒸发皿中,然后将蒸发皿推入硫分析仪的燃烧管中。对于液体校准物质和样品,在称取样品前,用约2g海砂覆盖蒸发皿的底部。称取的样品用另一部分海砂(约1g)覆盖。应选择校准物质和样品量并相互匹配,从而称取的量尽可能在0.1-0.2g范围内。样品重量应为约150mg。对于颗粒状样品,在测量前均质化该材料(例如,通过粉碎、研钵和研杵等)。
[0123]
应在测试样本的预期测量范围内选择校准的标准起始重量。使用前,标准品必须在105℃下干燥2小时。根据样品的预期硫含量,选择标准品:
[0124]
对于硫不超过2.5%的测试样本的校准,使用以下校准物质:
[0125]
502-671煤炭标准品,约1%的硫;来自leco
[0126]
502-422白油标准品,约2%的硫;来自leco
[0127]
对于硫超过2.5%的测试样本的校准,使用以下校准物质:
[0128]
对氨基苯磺酸(sulfanilic acid)标准品,来自eltra
[0129]
通过计算包含至少4个校准点的校准线,完成校准。使用leco仪器软件对测试结果进行评价和计算。
[0130]
d75
mod
或mode
mod
–
利用圆盘离心机,测定通过超声能量解聚后改性二氧化硅的粒径分布
[0131]
将待分析的样品分散在丙烷-1,2-二醇/水混合物中,用超声能量解聚,随后在圆盘离心机中根据粒径进行分离:在cps instruments的dc24000圆盘离心机上进行分析。
[0132]
将圆盘离心机设定为速度为20 000rpm。
[0133]
通过制备,用加入丙烷-1,2-二醇和nonidet(nonidet p40,来自applichem gmbh)的蔗糖溶液的密度梯度填充运行的圆盘离心机,并用十二烷顶层覆盖。
[0134]
本文的糖溶液的浓度在w=8%和w=24%蔗糖之间。密度梯度分为10级:
[0135]
24.0%/22.2%/20.4%/18.7%/16.9%/15.1%/13.3%/11.6%/9.8%/8.0%。
[0136]
为此,制备了两种具有以下组成的糖溶液:
[0137]
高密度溶液:24%蔗糖水溶液+0.2%nonidet
[0138]
低密度溶液:8%蔗糖水溶液+24%丙烷-1,2-二醇+0.2%nonidet
[0139]
将用于每个密度级的1.8ml的糖溶液注入圆盘离心机中,从最高浓度开始。为此,
在剂量注射器中以所需比例将两种糖溶液相互混合,如下所示:
[0140][0141][0142]
最后,注入0.5ml的十二烷。
[0143]
称取0.10g(
±
0.01g)样品,置于30ml的快盖小瓶(snap-lid vial)中,并加入15ml由24%丙烷-1,2-二醇+0.2%nonidet在软化水中组成的溶液。
[0144]
在夹子的辅助下,将填充好的快盖小瓶固定在冷却浴(julabo f12)中。冷却浴已预平衡至5℃(
±
1℃)。放置超声探头(来自hielscher;具有s14超声电极的up200s),从而超声电极浸没在小瓶中5.5cm处,从快盖小瓶的上边缘测量。样品在100%功率下连续超声处理15min。
[0145]
程序
[0146]
测量时,应在仪器的专用软件中建立以下参数:
[0147]
样品参数(软件描述中的小数位以点表示,而非逗号):
[0148]
最大直径:5.0微米
[0149]
最小直径:0.02微米
[0150]
颗粒密度:2.11g/ml
[0151]
颗粒折射率:1.46
[0152]
颗粒吸收:0.001k
[0153]
非球面系数:1.0
[0154]
校准标准品参数:
[0155]
峰直径:xxx微米1)
[0156]
半高峰宽:0.15微米
[0157]
颗粒密度:1.385g/ml 1
)
[0158]
流体参数:
[0159]
流体密度:1.075g/ml
[0160]
流体折射率:1.3706
[0161]
流体粘度:2.0cps
[0162]
呈现参数:
[0163]
显示模式:重量
[0164]
x轴标度:对数
[0165]
注:1)-根据使用的校准标准品,对应于制造商的说明书进行输入(例如pvc参比标准品,来自cps instruments)。
[0166]
测量波长设定为470nm。
[0167]
每种情况下,注入0.1ml标准品或样品悬浮液。对各待分析的样品进行双重测定(包括分散)。
[0168]
结果
[0169]
原始数据曲线(吸收测量)用于通过仪器的专用软件确定重量分布。现报告如下:
[0170]
mode
mod
(单位为nm)-最常见的粒径,对应于对数分布函数的最大值的横坐标值;
[0171]d75,mod
(超75重量%的颗粒的尺寸,单位为nm)-粒径分布的75%高于
[0172]
其值时的粒径(来自累积曲线);
[0173]
d75
mod
或mode
mod
–
通过圆盘离心机,采用超声能量测定解聚后二氧化硅的粒径分布
[0174]
将待分析的样品在水性悬浮液中用超声能量解聚,随后在圆盘离心机中根据粒径进行分离:在cps instruments的dc24000圆盘离心机上进行分析。
[0175]
以类似于改性二氧化硅方法的方法,在20 000rpm转速下进行测定。
[0176]
有偏差时,使用以下浓度的糖溶液(w=8%和w=24%蔗糖之间)分9级建立密度梯度:
[0177]
24%/22%/20%/18%/16%/14%/12%/10%/8%
[0178]
将用于各密度级的1.6ml糖溶液注入圆盘离心机中,从最高浓度开始。最后,注入0.5ml的十二烷。
[0179]
测定前,样品用研磨机(fritsch研磨机;具有80μm筛的pulverisette 14)进行超细研磨。称取0.75g(
±
0.05g)的研磨材料,置于30ml的快盖小瓶中,并加入15ml的软化水。
[0180]
偏离时,在仪器的专用软件中设置以下参数以用于测量:
[0181]
样品参数(软件描述中的小数位以点表示,而非逗号):
[0182]
最大直径:5.1微米
[0183]
最小直径:0.02微米
[0184]
颗粒密度:2.0g/ml
[0185]
颗粒折射率:1.44
[0186]
颗粒吸收:0.001k
[0187]
非球面系数:1.1
[0188]
校准标准品参数:
[0189]
峰直径:xxx微米2)
[0190]
半高峰宽:0.2微米
[0191]
颗粒密度:1.385g/ml 1
)
[0192]
流体参数:
[0193]
流体密度:1.045g/ml
[0194]
流体折射率:1.344
[0195]
流体粘度:1.2cps
[0196]
呈现参数:
[0197]
显示模式:重量
[0198]
x轴标度:对数
[0199]
2)
取决于特定的校准标准品。
[0200]
评价类似于改性二氧化硅的方法。
[0201]
干燥损失
–
根据din en iso 787-2测定干燥损失
[0202]
测定在干燥箱中于105℃下加热2h的样品的重量损失。
[0203]
该程序符合din iso 787-2及下述的说明:
[0204]
称量瓶(带凸缘盖;直径约80mm,高度约30ml)盖上盖子,在105℃下加热约1h。在干燥器中冷却后,插入盖子。在精密天平上精确测定重量至0.01g。精确称取5-10g的样品(重量取决于堆密度),以均匀层铺在称量瓶的底部。小心打开称量瓶,在干燥柜中于(105
±
2)℃下加热2h(盖子也加热)。
[0205]
小心用盖密封称量瓶,置于干燥器中冷却,并精确重新称重至0.01g。
[0206]
计算
[0207][0208]
e=起始重量,单位为g
[0209]
a=最终重量,单位为g
[0210]
ph
–
根据din en iso 787-9的二氧化硅的ph
[0211]
a.未改性二氧化硅的测定方法
[0212]
该程序符合din iso 787-9及下述的说明:
[0213]
称重前,用研钵和研杵将颗粒样品材料粉碎。
[0214]
制备待分析的样品的5%(m/m)水性悬浮液。为此,使用软化水(dmpa)。
[0215]
测量ph值前,在搅拌器上搅拌样品悬浮液至少5分钟。
[0216]
在之前校准的ph计(metrohm,型号780,带有ph电极6.0228.000(metrohm)),测量ph值。
[0217]
b.改性疏水二氧化硅的测定方法
[0218]
该程序符合din iso 787-9,说明及偏离如下所述:
[0219]
称重前,用研钵和研杵将颗粒样品材料粉碎。
[0220]
称取5g的待分析的样品。向其中加入40ml的软化水(dm)和60ml的甲醇p.a。
[0221]
测量ph值前,在搅拌器上搅拌样品悬浮液至少5分钟。
[0222]
在之前校准的ph计(metrohm,型号780,带有ph电极6.0228.000(metrohm)),测量ph值。
[0223]
c.改性亲水性二氧化硅的测定方法
[0224]
该程序符合din iso 787-9及下述的说明:
[0225]
称重前,用研钵和研杵将颗粒样品材料粉碎。
[0226]
制备待分析的样品的5%(m/m)水性悬浮液。为此,使用煮沸的软化水(dmpa)。
[0227]
测量ph值前,在搅拌器上搅拌样品悬浮液至少5分钟。
[0228]
在之前校准的ph计(metrohm,型号780,带有ph电极6.0228.000(metrohm)),测量
ph值。
[0229]
tar
–
通过未改性二氧化硅的脆性试验测定的颗粒磨损
[0230]
对于通过颗粒脆性试验测定的磨损,除去二氧化硅的细级分和粗级分,并使用3.15-5.00mm的级分。颗粒级分在旋转脆性室(例如erwekatar220,左右带有脆性鼓)中受到重复机械应力30分钟。随后,利用500μm筛除去所得细级分。质量差异(单位为%)对应于颗粒磨损。
[0231]
磨损测定重复进行两次。通过小心地手动筛分(带有金属筛网的分析筛,iso 3310-1,筛孔直径200mm
–
标称筛孔尺寸500μm、3.15mm和5.00mm,以及筛盘),筛分出3.15-5.00mm的颗粒级分。这排除了细颗粒和非常粗的颗粒。在分析天平或精密天平上精确称取(5.00
±
0.1)g的3.15-5.00mm级分。必须采集样品的代表性部分。将样品加入脆性鼓中;将其安装在磨损试验机上。该仪器在65rpm下运行30分钟。随后,将材料定量施加至500μm筛上,并通过适度搅拌/移动3-5秒除去粘附的细颗粒级分。然后在精密天平或分析天平上重新精确称重颗粒至0.01g。
[0232]
评价
[0233][0234]
磨损:颗粒脆性测试的磨损,单位为%
[0235]
e:筛分出的3.15-5.00mm(或1.00-3.15mm)级分的起始重量,单位为g
[0236]
a:施加应力和去除细颗粒后的残渣,单位为g
[0237]
测量结果是两次单独测量的平均值,并以%报告,保留一位小数。
[0238]
tar
mod
–
通过改性二氧化硅的脆性试验测定的颗粒磨损
[0239]
类似于上述未改性二氧化硅的tar测定,但是,除去二氧化硅的细级分和粗级分,并使用1.00-3.15mm级分。
[0240]
灼烧残渣
–
根据iso 3262-1或astm d 6740测定二氧化硅的灼烧残渣
[0241]
a.未改性二氧化硅的测定方法(iso 3262-1)
[0242]
通过在1000℃下于点火炉中灼烧沉淀的二氧化硅2h,测定总含水量(物理的和化学结合的),从而测定所有挥发性组分(灼烧损失)的含量,也可据此计算灼烧残渣。
[0243]
测定程序
[0244]
在精确度为
±
0.1mg的分析天平上,借助刮刀,在两个瓷坩埚或熔融坩埚中在每种情况下称取约500mg的二氧化硅。随后,将坩埚连同二氧化硅一起在(1000
±
50)℃的点火炉中灼烧(120
±
5)min。
[0245]
灼烧后,将坩埚置于具有合适干燥剂的干燥器中冷却约1.5-2.0h,并用分析天平重新称重。重复进行两次测定。
[0246]
评价
[0247]
首先,根据干燥物质计算灼烧损失(il
(干燥)
):
[0248]
[0249]
il
(干燥)
:基于105℃下干燥2h的物质的灼烧损失 单位为%
[0250]
me:称取的二氧化硅质量 单位为g
[0251]
ma:灼烧后二氧化硅的质量 单位为g
[0252]
dl:105℃下2h的干燥损失 单位为%
[0253]
根据基于原始物质的灼烧损失(il
(原始)
),如下计算基于原始物质的灼烧残渣(ir
(原始)
):
[0254]
il
(原始)
=il
(干燥)
*(100-dl)/100(单位为%)
[0255]
因此
[0256]
ir
(原始)
=100%-dl-il
(原始)
(单位为%)
[0257]
为此,通过“根据din en iso 787-2测定干燥损失”的方法测定干燥损失(见上文)。
[0258]
b.改性二氧化硅的测定方法(astm d 6740)
[0259]
残渣主要由sio2组成,其形成于硅烷分解和二氧化硅灼烧过程中。该方法符合astm d 6740。
[0260]
称取约(0.5-1.0)g1的待分析的样品,置于预先煅烧的坩埚中,精确度为0.1mg。加入约2ml的90%硫酸,小心倾斜坩埚以进行混合。
[0261]
将坩埚置于初步灰化设备(型号:svr/e,可控至2500瓦)中,并从10%(=250瓦)开始逐渐加热。除去酸后(样品几乎干燥),加热初步灰化设备至最高温度。随后,将坩埚置于预热的马弗炉(1000℃)中2h。如果灰分此后仍为灰色或黑色,则在1000℃下进一步灼烧2h。将坩埚置于干燥器中冷却,精密称重至0.1mg。
[0262]
_________________________
[0263]1应选择起始重量,使得残渣质量大于100mg。
[0264]
计算
[0265]
灼烧残渣r计算如下:
[0266][0267]
m1=样品重量,单位为g
[0268]
m2=残渣质量,单位为g
[0269]
(灼烧损失v(单位为%)表示为v=100-r)
[0270]
pv(v80,3.7-80nm,140
°
),if(140
°
,dv/dr),is(90%,140
°
,dv/dr),r
min
(90%,140
°
,dv/dr)-根据din 66133,基于压汞法测定孔隙半径和孔隙体积
[0271]
在0.03至420mpa的压力范围内,确定了加压二氧化硅样品的孔径、相应的孔隙体积和孔隙分布。根据din 66133使用micromeritics autopore iv9520进行测定。
[0272]
在偏离din 66133中,在测量前对二氧化硅样品进行压力处理。这是使用specac atlas手动15吨液压机完成的。在specac ltd.的内径为13mm的压片模中加入1g样品,并根据显示加压1t。这一负载保持1min。然后将样品减压,并在105
±
2℃的干燥箱中干燥2h。
[0273]
对于测量,称取制备的样品,置于micromeritics 16型透度计(penetrometer)中。称取约330mg,精确到0.001g。随后,在测量仪器的低压端口中将透度计逐渐抽空至50μm的hg,并在该压力下放置5min。随后,先在低压端口,再在高压端口为透度计充汞至420mpa,记
录测量曲线(压力/体积曲线)。autopore仪器按照micromeritics的操作说明进行操作,并由软件控制。每次测量均通过透度计的空白测量进行校正。总体测量范围为0.003-420mpa。
[0274]
根据din 66133,使用标准值,由测量曲线计算测量结果:
[0275]
接触角:140
°
[0276]
表面张力:480dyn/cm
[0277]
评价以下参数:
[0278]
pv(v80,3.7-80nm,140
°
)
[0279]
if(140
°
,dv/dr)
[0280]
is(90%,140
°
,dv/dr)
[0281]rmin
(90%,140
°
,dv/dr)
[0282]
ro-tap(》150μm;》300μm;》500μm)
–
按照iso 5794-1附录f进行筛分分析
[0283]
使用旋转筛分机(tyler ro-tap rx-29分析筛分机,带有计时器开关),进行筛分分析。该方法按照iso 5794-1附录f进行。对于筛分分析,将具有不同筛孔尺寸的试验筛叠放在一起(带金属筛网的分析筛,iso 3310-1,标称筛孔尺寸150μm,筛孔直径200mm;带金属筛网的分析筛,iso 3310-1,标称筛孔尺寸300μm,筛孔直径200mm;带金属筛网的分析筛,iso 3310-1,标称筛孔尺寸500μm,筛孔直径200mm)。筛分5分钟后,称重相应级分。
[0284]
筛分塔安装在ro-tap筛分机中。自下而上的顺序如下:筛盘、150μm筛、300μm筛和500μm筛。测定前轻轻均质化样品。在精密天平上称取(100
±
1)g,精确至0.01g,置于烧杯中,并将该样品定量转移至最上面的筛(500μm)上。使用敲击器(tapper)进行ro-tap筛分(5min
±
5s)。筛分结束后,移除筛分塔,称重150μm、300μm和500μm筛上的级分。
[0285]
计算
[0286]
ro-tap》500μm(单位为%)=(a
500
·
100%)/e
[0287]
以及
[0288]
ro-tap》300μm(单位为%)=(a
300
·
100%)/e
[0289]
和
[0290]
ro-tap》150μm(单位为%)=(a
150
·
100%)/e
[0291]a500
=500μm筛上的残渣,单位为g
[0292]a300
=300μm筛上的残渣,单位为g
[0293]a150
=150μm筛上的残渣,单位为g
[0294]
e=起始重量,单位为g
[0295]
与iso 5794-1附录f的偏离
[0296]
在与标准的偏离中,根据该检测说明,使用筛孔尺寸为150μm、300μm和500μm的筛。
[0297]
doa吸收
–
根据iso 19246测定doa吸收
[0298]
对于性能,将12.50
±
0.02g样品引入具有扩展功能/评价单元的brabender absorptometer e的捏合器腔室中。然后,通过恒定揉捏,以4ml/min的计量速率计量加入己二酸二辛酯(例如doa)。捏合器速度为125rpm。该程序使用原始数据曲线计算多项式(polynomial)。该多项式的最大扭矩的70%值用于确定基于原始材料的doa吸收,单位为ml/(100g)。根据iso 19246进行测定。
[0299]
对于二氧化硅颗粒,使用1.00-3.15mm颗粒级分进行测定,该级分必须通过使用适
当的筛网进行筛分事先制备。
[0300]
应在测量仪器软件中进行以下设置:
[0301]
试验条件
[0302]
计量速度(滴定管):4.0ml/min
[0303]
捏合器速度:125min-1
[0304]
温度:23.0℃
[0305]
评价
[0306]
实验结束
[0307]
扭矩阈值:100mnm
[0308]
结束:最大值后60s
[0309]
扭矩极限:10000mnm
[0310]
多项式
[0311]
开始:最大扭矩的50%
[0312]
结束:最大值后20s
[0313]
使用具有不同doa吸收的适当参比材料,可以借助软件,对分析捏合器进行单独标准化。基于已确定的标准化函数(线性方程y=a*x+b),基于原始物质(单位为ml/(100g)),doa吸收(标准化)表示为最大扭矩值的70%,摘自测量报告。
实施例
[0314]
使用的参照二氧化硅为以下二氧化硅或改性二氧化硅(表1)。单位phf(填料百分数)是指基于100重量份的二氧化硅,添加剂的重量份:
[0315]
二氧化硅1是来自evonikresourceefficiencygmbh的9100gr。
[0316]
二氧化硅2根据ep1525159b1中的实施例1制备。
[0317]
二氧化硅3是来自evonikresourceefficiencygmbh的7000gr。
[0318]
二氧化硅4是来自solvay的premium200mp。
[0319]
二氧化硅5是来自solvay的1165mp。
[0320]
二氧化硅6根据ep0901986b1中的实施例4制备。
[0321]
二氧化硅7是来自ppgindustriesohio,inc.的ciptane
tm
。
[0322]
二氧化硅8+9是来自ppgindustriesohio,inc.的400和458。
[0323]
二氧化硅10根据wo2014033300a1中的实施例1制备。
[0324]
二氧化硅11是来自evonikresourceefficiencygmbh的8113gr。
[0325]
二氧化硅12基于ep0901986b1的实施例4,用5phfsi改性。
[0326]
二氧化硅13基于ep0901986b1的实施例4,用10phfsi改性。
[0327][0328]
通过在输送螺杆中初步混合起始二氧化硅和添加剂,然后在henschel混合器(thyssen的henschel fm 40流体混合器)中干燥,制备本发明的二氧化硅14-21。对于实施
例14-21,使用了根据ep 0901986 b1的实施例4制备的3000g起始二氧化硅。根据配方使用添加剂(表2)。将混合器预热至100℃。加入二氧化硅/添加剂混合物后,在henschel混合器中以2500rpm转速干燥4min。
[0329]
在实施例14、16、18和20中,然后在碾压机中压实改性二氧化硅。
[0330]
表2显示了根据发明的改性二氧化硅的组合物。单位phf(填料百分数)是指基于100重量份的二氧化硅,添加剂的重量份:si是evonik resource efficiency gmbh的双[(3-三乙氧基甲硅烷基)丙基]二硫化物。nxt是momentive的(eto)3si-(ch2)
3-s-c(o)-c7h
15
。
[0331]
表2
[0332][0333]
表3显示了根据发明的改性二氧化硅的分析数据。
[0334][0335]
在旋转闪蒸干燥器中制备根据本发明的改性二氧化硅22-29。根据ep0901986b1的实施例4和ep 1525159 b1的实施例1,制备用于改性的基础二氧化硅。通过输送螺杆将所得
滤饼输送至旋转闪蒸干燥器中。在将所得混合物计量至干燥室之前,通过导管将添加剂添加至输送单元中。任选地对干燥的二氧化硅进行造粒。
[0336]
使用的硅油为dow xiameter
tm
pmx-200silicone fluid聚二甲基硅氧烷,其粘度为50cst。
[0337]
表4显示了改性二氧化硅的分析数据。
[0338]
表4
[0339][0340]
橡胶特性检查
[0341]
用于橡胶混合物的材料列于表5中。使用的另一种参照二氧化硅为evonik resource efficiency gmbh的vn 3gr。
[0342]
配方见表6。
[0343]
表5:实施例中使用的材料列表
[0344][0345]
表6:s-sbr/br混合物的混合物配制物
[0346]
混合物1234对照5对照6对照7阶段1
ꢀꢀꢀꢀꢀꢀꢀ
ssbr96.396.396.396.396.396.396.3br30303030303030本发明的改性二氧化硅1590.6
‑‑‑‑‑‑
本发明的改性二氧化硅21-91.4
‑‑‑‑‑
本发明的改性二氧化硅19
‑‑
90.6
‑‑‑‑
本发明的改性二氧化硅17
‑‑‑
90.6
‑‑‑
ultrasil vn 3gr
‑‑‑‑
80
‑‑
二氧化硅5
‑‑‑‑‑
80-二氧化硅6
‑‑‑‑‑‑
80si 266
‑‑‑‑
6.46.46.4zno2.02.02.02.02.02.02.0硬脂酸2.02.02.02.02.02.02.0油8.758.758.758.758.758.758.75蜡2.02.02.02.02.02.02.0ppd2.02.02.02.02.02.02.0tmq1.51.51.51.51.51.51.5阶段2
ꢀꢀꢀꢀꢀꢀꢀ
阶段1批料
ꢀꢀꢀꢀꢀꢀꢀ
dpg2.52.52.52.52.52.52.5阶段3
ꢀꢀꢀꢀꢀꢀꢀ
阶段2批料
ꢀꢀꢀꢀꢀꢀꢀ
cbs1.61.61.61.61.61.61.6硫2.142.22.142.142.142.142.14tbztd0.20.20.20.20.20.20.2
[0347]
使用harburg freudenberger maschinenbau gmbh的gk 1.5e密闭式混合机,制备橡胶混合物(表7)。
[0348]
表7:s-sbr/br混合物的混合物制备
[0349][0350][0351]
本文指定的橡胶混合物或其硫化产物的物理试验结果列于表8中。硫化产物是由第3阶段未处理的混合物在130bar下165℃下加热15min制备的。通过表9中所述的方法对橡胶混合物进行测量。
[0352]
表8:本文指定的橡胶混合物及其硫化产物的物理试验结果
[0353][0354]
从表8可以明显看到,与参照混合物(5-7)相比,根据本发明的化合物物(1-4)具有改善的加工特性,表现为门尼粘度减小(混合阶段2和3)和最小扭矩ml减小(混合阶段3)。混合阶段3后转化t10%和t90%的时间也证明了具有优化的硫化转化的扩展处理窗口。此外,与参照混合物(5-7)相比,根据本发明的混合物(1-4)在具有相同抗拉强度的情况下显示出改善的断裂伸长率。根据本发明的硫化混合物的动态性能优于参照(5-7),同时具有良好的耐磨性(混合物1、3和4)和分散质量。
[0355]
表9:
[0356]
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1