一种利用荧光碳量子点制备纳米硒的方法和应用
1.本发明属于纳米材料技术领域,特别涉及一种利用荧光碳量子点制备纳米硒的方法和应用。
背景技术:
2.碳量子点是一类尺寸在10nm以下,带有sp2/sp3杂化碳的无定型或纳米晶核结构的新型零维碳纳米材料,不仅具有传统量子点优异的光学性能,同时还具有易于合成和功能化、成本低廉、原材料广泛、毒性低和生物相容性好等显著特点,广泛应用于生物成像、化学传感、药物传递、光催化和太阳能电池等领域。一般地,碳量子点表面富含的-oh、-cooh等亲水性基团及c=o、c=c,可为其表面修饰提供良好的条件。经过表面钝化或杂原子掺杂等表面修饰的碳量子不仅可调节和增强其发光性能,还可提高其生物相容性、光稳定性、靶向性等,使其更好地应用于生物、化学传感分析等领域。
3.纳米硒是平均直径为1~100nm的无定型红色单质硒,具有多种生物学功能和较高生物可及性,可用作生物体内抗氧化剂和抗癌剂。同时,硒作为人体必需微量元素之一,对人类健康至关重要。与无机硒、有机硒相比,纳米硒具有高效、低毒特性,可作为新型的硒补充剂。故制备稳定性高、功能活性强的纳米硒成为硒研究的重点和热点。但是,纳米硒极不稳定,受热易转变为热力学稳定的三方晶系t-se而失去生物活性,通常需要加入模板作为稳定剂。软模板法是目前制备纳米硒最主要的方法之一,它是利用富含羟基、羧基、氨基、碳基等官能团的蛋白质、多糖、多酚及其他生物大分子与纳米硒的相互作用使其稳定的同时,利用具有一定功能特性的软模板,能更进一步强化纳米硒的功能。
4.目前,人们致力于寻找具有一定生物学功能的软模板,以制备得到高稳定性和生物学功能强化型纳米硒。为进一步拓展、深化纳米硒的研究,本发明选用荧光碳量子点作为新型功能化软模板,成功建立了稳定性高、荧光性能强的无定型红色荧光碳量子点-纳米硒的制备方法,同时,基于硒和汞的高亲和力一步形成的硒化汞具有类氧化酶特性,成功构建基于荧光碳量子点-纳米硒荧光-比色双信号光学传感器,应用于汞离子的定量检测,成功克服了现有技术制备得到的纳米硒功能单一的缺点,并成功将纳米硒应用在化学传感分析等新领域,为制备新型纳米硒及开拓纳米硒应用新领域提供了新方法和新思路。
技术实现要素:
5.为了克服现有技术中存在的缺点和不足,本发明的首要目的在于提供一种利用荧光碳量子点制备纳米硒的方法;该方法先是以含碳物质为碳源,选用有机小分子或聚合物为表面钝化剂,含氮、磷、硫、硼化合物为掺杂原子化合物,经微波或水热碳化、裂解合成表面富含亲水性官能团、荧光性能强的荧光碳量子点;然后以富含羟基、羧基等亲水性官能团的荧光碳量子点为功能化软模板,采用软模板法制备得到具有良好荧光性能的荧光碳量子点-纳米硒;经证实荧光碳量子点具备稳定、分散并功能化纳米硒的能力。
6.本发明的另一目的在于提供一种上述方法制备得到的纳米硒。
7.本发明的再一目的在于提供一种基于上述荧光碳量子点-纳米硒的荧光-比色双信号构建的光学传感器检测hg
2+
的方法;本发明发现基于硒和汞的高亲和力一步形成的硒化汞可触发荧光碳量子点-纳米硒的类氧化酶活性,使其同时具备荧光、比色双信号;在hg
2+
存在下,基于硒和汞的高亲和力,触发荧光碳量子点-纳米硒的类氧化酶活性,成功构建基于荧光碳量子点-纳米硒的荧光-比色双信号光学传感器,实现hg
2+
的高选择、高灵敏的荧光-比色双信号检测;以荧光碳量子点-纳米硒为荧光探针,可实现对hg
2+
进行荧光定量检测;基于hg
2+
触发荧光碳量子点-纳米硒的类氧化酶活性实现对hg
2+
的比色定量检测。
8.本发明的又一目的在于提供一种上述荧光碳量子点-纳米硒的应用,所得荧光碳量子点-纳米硒可应用于生物学、光学、电学、磁学及催化等领域。
9.本发明的目的通过下述技术方案实现:
10.一种荧光碳量子点-纳米硒的制备方法,包括以下步骤:
11.(1)将碳源和表面修饰剂按10:1~1:5质量比溶解于超纯水中,所述表面修饰剂为表面钝化剂和/或掺杂原子的化合物;在微波条件下或直接加热、加压条件下得到表面修饰后的荧光碳量子点,超纯水重悬浮,直接收集或经离心得到上清液,将上清液过滤,直接获得或经透析后获得荧光碳量子点溶液;
12.(2)将荧光碳量子点、硒前体物质和还原剂的混合物溶液于20~100℃水浴静置反应0.5~24h,可直接获得或经透析分离后获得荧光碳量子点-纳米硒悬液;硒前体物质和还原剂的摩尔比为1:2~1:20。
13.步骤(1)所述碳源为柠檬酸、柠檬酸钠、抗坏血酸、抗坏血酸钠、葡萄糖,或上述物质任意比例的混合物;所述表面钝化剂为聚乙二醇(peg)、β-环糊精、十二烷基硫酸钠(sds),或上述物质任意比例的混合物;所述掺杂原子的化合物为氮掺杂化合物、磷掺杂化合物、硫掺杂化合物、硼掺杂化合物,或上述物质任意比例的混合物,包括尿素、乙二胺四乙酸二钠、磷酸、硼酸、l-半胱氨酸。
14.步骤(1)所述微波的功率为80~800w,微波处理时间为5~30min;
15.所述直接加热、加压采用的是水浴加热、油浴加热或高压反应釜加热,具体加热温度为100~350℃,加热时间为1~24h,压强为0~10mpa;
16.所述离心是在4~25℃条件下以转速27~12851
×
g离心2~40min;所述过滤是将上清液过0.1~0.22μm滤膜;所述透析是采用mwco 500~1000da透析袋透析12~72h。
17.步骤(2)所述硒前体物质为亚硒酸钠、亚硒酸、二氧化硒、硒酸或硒酸钠;所述还原剂为抗坏血酸、抗坏血酸钠、还原性糖、多酚类物质、水合肼或硼氢化钠;
18.所述硒前体物质在混合物溶液中的浓度为1~50mm,所述还原剂在混合物溶液中的浓度为2~400mm;所述荧光碳量子点在混合物溶液中的浓度为25~2000mg/l;
19.所述透析分离是采用mwco 1000~10000da再生纤维素透析袋透析6~72h。
20.更加优选的,步骤(1)所述碳源为柠檬酸;所述表面钝化剂为β-环糊精;所述掺杂原子的化合物为尿素;
21.更加优选的,步骤(2)所述硒前体物质为亚硒酸钠,亚硒酸钠在混合物溶液中的浓度为4mmol/l;所述还原剂为抗坏血酸,抗坏血酸在混合物溶液中的浓度为24mmol/l;所述反应时间为0.5h;所述反应温度为25℃;所述透析分离是采用mwco 8000da再生纤维素透析袋透析24h。
22.一种由上述的制备方法制备得到的荧光碳量子点-纳米硒。
23.一种基于上述的荧光碳量子点-纳米硒的荧光-比色双信号构建的光学传感器检测hg
2+
的方法,该方法包括以下两种方式;
24.a、所述基于荧光碳量子点-纳米硒荧光信号检测hg
2+
具体按照以下步骤:在浓度为0.2mol/l、ph为5.0的缓冲液条件下,浓度为0~800μmol/l的hg
2+
于20~80℃水浴下淬灭荧光碳量子点-纳米硒荧光2~30min,其荧光强度变化与hg
2+
浓度呈现良好线性关系;
25.b、所述基于hg
2+
触发荧光碳量子点-纳米硒的类氧化酶活性比色检测hg
2+
具体按照以下步骤:将浓度为0.94~120.00μg/ml的荧光碳量子点-纳米硒悬液与25~50nmol/l的hg
2+
在浓度为0.2mol/l、ph 2~10的缓冲液体系中作用2~20min;然后将混合体系与胺类底物在20~60℃水浴静置反应5~60min,生成有色物质,测定吸光度,计算得到的类氧化酶活性的吸光绝对值比值与hg
2+
浓度呈现良好线性关系。
26.方式a所述水浴的温度为25℃,淬灭反应的时间为20min;所述hg
2+
浓度在0.78~12.50μmol/l范围内与荧光碳量子点-纳米硒的荧光强度呈良好线性关系,检出限为0.14μmol/l。
27.方式b所述缓冲液体系为醋酸-醋酸钠缓冲液体系、柠檬酸-柠檬酸钠缓冲液、磷酸缓冲液;所述胺类底物为3,3’,5,5
’‑
四甲基联苯胺(tmb)、2.2-联氮-二(3-乙基-苯并噻唑-6-磺酸)二铵盐(abts)、邻苯二胺(opd)。
28.方式b所述荧光碳量子点-纳米硒悬液的浓度为15μg/ml,所述作用的时间为5min,所述缓冲液体系为ph为5,所述胺类底物为3,3’,5,5
’‑
四甲基联苯胺(tmb),所述水浴静置反应温度为30℃,所述水浴静置反应时间为10min,检出限为6.13nmol/l。
29.上述的荧光碳量子点-纳米硒在生物领域、检测领域和催化领域中的应用。
30.通过动态光散射和激光多普勒测速技术测定荧光碳量子点-纳米硒的dh、总光强、pdi和zeta电位,评价本发明所得荧光碳量子点-纳米硒的胶体化学特性;通过透射电子显微镜、扫描电子显微镜、傅里叶红外光谱、x射线衍射仪、x射线光电子能谱等表征荧光碳量子点-纳米硒的形态形貌、表面官能团组成和结构、晶型结构等,评价本发明所得荧光碳量子点-纳米硒的基本特性;分别通过紫外-可见吸收光谱和荧光光谱测定其物质组成特征吸收峰和荧光强度,评价本发明所得荧光碳量子点-纳米硒的光学特性和酶学特性。
31.本发明相对于现有技术具有如下的优点及有益效果:
32.本发明通过以富含羟基、羧基、氨基等亲水性官能团和荧光性能强的荧光碳量子点为新型功能化软模板,成功合成荧光碳量子点-纳米硒,赋予纳米硒以荧光碳量子点荧光等新型特性;同时发现,hg
2+
存在下,基于硒和汞的高亲和力可触发荧光碳量子点-纳米硒的类氧化酶活性,成功构建基于荧光碳量子点-纳米硒的荧光-比色双信号光学传感器,应用于化学传感分析领域,实现hg
2+
的高选择、高灵敏的荧光-比色双信号检测。本发明拓宽了纳米硒的研究思路,扩大了荧光碳量子点和纳米硒的应用范围,同时为制备稳定、分散的荧光纳米材料提供新思路。
附图说明
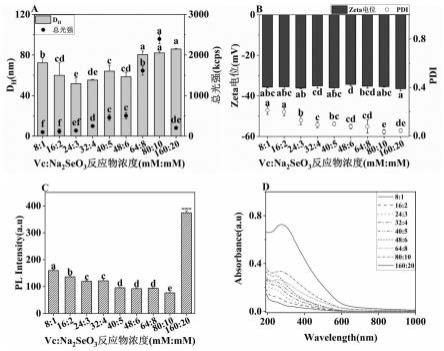
33.图1为不同vc与na2seo3浓度制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和uv-vis吸收光谱(d)。
34.图2为不同荧光碳量子点添加量制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和uv-vis吸收光谱(d)。
35.图3为不同vc与na2seo3配比制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和uv-vis吸收光谱(d)。
36.图4为不同反应时间制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和uv-vis吸收光谱(d)。
37.图5为不同反应温度制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和uv-vis吸收光谱(d)。
38.图6为不同碳源合成的碳量子点制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和类氧化酶活性的吸光值(d)。
39.图7为不同表面钝化剂修饰的碳量子点制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和类氧化酶活性的吸光值(d)。
40.图8为不同杂原子表面修饰的碳量子点制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和类氧化酶活性的吸光值(d)。
41.图9为水热合成的共掺杂柠檬酸碳量子点制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和类氧化酶活性的吸光值(d)。
42.图10为微波或水热合成的共掺杂抗坏血酸碳量子点制备的荧光碳量子点-纳米硒的dh、总光强(a)、pdi、zeta电位(b)、荧光强度值(c)和类氧化酶活性的吸光值(d)。
43.图11为hg
2+
存在下,荧光碳量子点-纳米硒悬液催化底物tmb前后的uv-vis吸收图谱(a)及其吸光度时间变化曲线(b)。
44.图12为hg
2+
作用荧光碳量子点-纳米硒悬液的荧光光谱(a)、荧光强度的比值(b)和hg
2+
浓度与荧光碳量子点-纳米硒悬液荧光强度比值的线性关系图(c)。
45.图13为不同种类、浓度的金属离子作用荧光碳量子点-纳米硒悬液的荧光强度值比值。
46.图14为hg
2+
作用荧光碳量子点-纳米硒类氧化酶活性的吸光绝对值比值(a)和hg
2+
与荧光碳量子点-纳米硒类氧化酶活性吸光绝对值比值的线性关系图(b)。
47.图15为不同种类、浓度金属离子作用荧光碳量子点-纳米硒类氧化酶活性的吸光绝对值比值。
48.图16为荧光碳量子点-纳米硒与hg
2+
作用后的xps图谱。xps全谱扫描(a),c 1s分峰拟合图谱(b),se 3d分峰拟合图谱(c)和hg 4f分峰拟合图谱(d)。
具体实施方式
49.下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。为了使本发明的目的、技术方案更加清晰明确,下面结合以表面钝化、杂原子掺杂等表面修饰的荧光碳量子点为新型功能化模板举例,对本发明进行进一步详细说明。本发明所用设备仪器和试剂均为本领域所常用。应当理解,此处所描述的举例仅仅用以解释本发明,并不用于限定本发明。
50.实施例1:一种利用荧光碳量子点制备纳米硒的方法。
51.β-环糊精-氮掺杂荧光碳量子点(β-cd-n-cqds)的合成:称取β-环糊精、尿素和柠
檬酸各1g于100ml锥形瓶中,加入10ml超纯水溶解,置于微波炉中央,设置微波功率800w,加热5min,加入10ml超纯水复溶,于4℃、3824
×
g离心10min,上清液过0.22μmol/l滤膜,真空冷冻干燥,待用。
52.将一定量β-cd-n-cqds粉末(500mg/l)溶解于超纯水中,分别加入0.25、0.50、0.75、1.00、1.25、1.50、2.00、2.50、5.00ml的na2seo3溶液(100mmol/l),磁力搅拌5min,静置15min,分别加入0.4、0.8、1.2、1.6、2.0、2.4、3.2、4.0、8.0ml的vc溶液(500mmol/l),磁力搅拌5min,于40℃静置反应1h,于8000da再生纤维素透析袋透析24h(每2.5h换水一次)获得荧光碳量子点-纳米硒悬液,测定其dh、总光强、pdi、电位、uv-vis光谱和荧光强度,如图1所示,当vc:na2seo3浓度在32mmol/l:4mmol/l时,荧光碳量子点-纳米硒颗粒dh小、粒径分布集中,静电稳定性高,荧光发光强。故选择此浓度开展后续实验。
53.实施例2:一种利用荧光碳量子点制备纳米硒的方法。
54.与实施例1相比,区别点仅在于:
55.控制vc:na2seo3浓度为32mmol/l:4mmol/l,改变β-cd-n-cqds添加量为0、100、200、300、400、500、600、700、800、900mg/l,制备荧光碳量子点-纳米硒,测定其dh、总光强、pdi、电位、uv-vis光谱和荧光强度,如图2所示,当β-cd-n-cqds添加量为400mg/l时,荧光碳量子点-纳米硒的dh小、粒子粒径分布较集中,静电稳定性高、荧光发光强度高,故选择此添加量开展后续实验。
56.实施例3:一种利用荧光碳量子点制备纳米硒的方法。
57.与实施例1相比,区别点仅在于:
58.控制na2seo3浓度为4mmol/l,β-cd-n-cqds添加量为400mg/l,改变vc浓度为8、16、24、32、40、48mmol/l,制备荧光碳量子点-纳米硒,测定其dh、总光强、pdi、电位、uv-vis光谱和荧光强度,如图3所示,当vc:na2seo3浓度为24mmol/l:4mmol/l时,荧光碳量子点-纳米硒的dh小、粒子数目多、粒子粒径分布集中,zeta电位值和荧光强度值最大,具有高的静电稳定性和发光强度。
59.实施例4:一种利用荧光碳量子点制备纳米硒的方法。
60.与实施例1相比,区别点仅在于:
61.控制vc:na2seo3浓度为24mmol/l:4mmol/l,β-cd-n-cqds添加量为400mg/l,改变反应时间为0.5、1.0、2.0、3.0、4.0、5.0h,制备荧光碳量子点-纳米硒,测定其dh、总光强、pdi、电位、uv-vis光谱和荧光强度,如图4所示,反应时间在0.5h时,荧光碳量子点-纳米硒的dh最小、zeta电位值和荧光强度值最大,表明在此反应条件下,反应0.5h即可充分完成反应,可制备得到粒子粒径小、数目多,静电稳定性高,荧光发光强度强的荧光碳量子点-纳米硒。
62.实施例5:一种利用荧光碳量子点制备纳米硒的方法。
63.与实施例1相比,区别点仅在于:
64.控制vc:na2seo3浓度为24mmol/l:4mmol/l,β-cd-n-cqds添加量为400mg/l,反应时间为0.5h,改变反应温度为20、25、40、60、80、100℃,制备荧光碳量子点-纳米硒,测定其dh、总光强、pdi、电位、uv-vis光谱和荧光强度,如图5所示,当反应温度为25℃时,荧光碳量子点-纳米硒的dh最小、zeta电位值和荧光强度值大,表明在25℃条件下,可制备得到粒子粒径小、分布集中,具有较高静电稳定性和较强发光强度的荧光碳量子点-纳米硒。
65.实施例6:一种利用荧光碳量子点制备纳米硒的方法。
66.与实施例1相比,区别点仅在于:
67.控制vc:na2seo3浓度为24mmol/l:4mmol/l,β-cd-n-cqds添加量为400mg/l,反应时间为0.5h,反应温度为25℃,制备荧光碳量子点-纳米硒。
68.实施例7:荧光碳量子点-纳米硒胶体化学特性、基本特性和光学特性表征。
69.采用动态光散射和激光多普勒测速等技术表征实施例6制备的荧光碳量子点-纳米硒的平均流体力学粒径和zeta电位,研究本发明所得荧光碳量子点-纳米硒的胶体化学特性。结果显示:荧光碳量子点-纳米硒具有良好的水溶性,呈深红色溶液,平均流体力学直径为50.76nm,总光强为202.07kcps,pdi为0.133,颗粒粒径呈单峰分布,分布均一,zeta电位为-45.67mv,具有高的静电稳定性。
70.采用透射电子显微镜、扫描电子显微镜、傅里叶红外光谱、x射线衍射仪、x射线光电子能谱等表征实施例6制备的荧光碳量子点-纳米硒的形态形貌、表面官能团组成和结构、晶型结构等,研究本发明所得荧光碳量子点-纳米硒的基本特性。结果显示:荧光碳量子点-纳米硒呈类球形结构,单分散性良好,粒子粒径大小分布均一,经高斯拟合得到荧光碳量子点-纳米硒实际平均直径为33.76
±
7.72nm,表面含有-oh、-nh2、-c=o等亲水性基团,为无定型零价纳米硒。
71.采用紫外-可见吸收光谱和荧光光谱测定实施例6制备的荧光碳量子点-纳米硒的物质组成特征吸收峰和荧光强度,研究本发明所得荧光碳量子点-纳米硒的光学特性。结果显示:荧光碳量子点-纳米硒的吸收主要集中在200~600nm范围内,在247nm处具有明显特征吸收峰,主要归因为碳核里c=c的π-π*的跃迁,其荧光发射属于激发依赖型,最大激发波长为426nm,最大发射波长为522nm。
72.实施例8:hg
2+
触发荧光碳量子点-纳米硒的类氧化酶活性。
73.将实施例6制备的荧光碳量子点-纳米硒溶液50μl与naac(0.2mol/l,ph 4.0)缓冲液412.5μl混合,加入2mmol/l hgcl2溶液12.5μl,静置反应5min,加入4g/l显色底物tmb溶液25μl,反应体系置于30℃恒温水浴静置反应20min,取出置于冰浴中30s,测定其uv-vis吸收光谱(扫描范围400-800nm),结果如图6所示,在hg
2+
存在下,荧光碳量子点-纳米硒可有效催化氧化tmb,在652nm处具有强吸收峰,表明hg
2+
可触发荧光碳量子点-纳米硒的类氧化酶活性。
74.实施例9:一种利用荧光碳量子点制备纳米硒的方法。
75.与实施例1相比,区别点仅在于:
76.荧光碳量子点的合成:分别以柠檬酸、柠檬酸钠、葡萄糖、抗坏血酸为碳源直接合成荧光碳量子点。
77.控制vc:na2seo3浓度为6mmol/l:1mmol/l、荧光碳量子点添加量为500mg/l,制备荧光碳量子点-纳米硒,测定其dh、总光强、pdi、电位、荧光强度和类氧化酶活性,结果如图7所示,荧光碳量子点-纳米硒的dh小、粒子数目多、粒子粒径分布集中;除葡萄糖外,其余碳源zeta电位值均大于30mv,具有高的静电稳定性;以柠檬酸、柠檬酸钠为碳源的荧光碳量子点-纳米硒荧光强度值较大,具有高的发光强度;以抗坏血酸为碳源的荧光碳量子点-纳米硒的类氧化酶活性吸光值最大,具有较高的类氧化酶活性,其余碳源的荧光碳量子点-纳米硒的类氧化酶活性较弱。
78.实施例10:一种利用荧光碳量子点制备纳米硒的方法。
79.与实施例9相比,区别点仅在于:
80.以柠檬酸为碳源,分别以β-环糊精、聚乙二醇(peg 4000)为表面钝化剂合成的荧光碳量子点为软模板,制备荧光碳量子点-纳米硒,其dh、总光强、pdi、电位、荧光强度和类氧化酶活性结果如图8所示。当以β-环糊精为表面钝化剂时,制备得到的荧光碳量子点-纳米硒的dh较小、粒子数目较少、粒子粒径分布集中;两种荧光碳量子点-纳米硒的zeta电位值均大于30mv,均具有高的静电稳定性;以peg 4000为表面钝化剂时,制备得到的荧光碳量子点-纳米硒的荧光强度值较大,具有高的发光强度;但以β-环糊精为表面钝化剂制备的荧光碳量子点-纳米硒类氧化酶活性吸光值更大,具有更强的类氧化酶活性。
81.实施例11:一种利用荧光碳量子点制备纳米硒的方法。
82.与实施例9相比,区别点仅在于:
83.以柠檬酸为碳源,分别以尿素(u)、乙二胺四乙酸二钠(edta-2na)、磷酸(pa)、硼酸(ba)、l-半胱氨酸(cys)为氮、磷、硼、硫原子掺杂剂合成的荧光碳量子点为软模板,制备荧光碳量子点-纳米硒,其dh、总光强、pdi、电位、荧光强度和类氧化酶活性结果如图9所示。除硼酸外,其余杂原子制备的荧光碳量子点-纳米硒dh小、粒子数目多、粒子粒径分布集中;除尿素和硼酸外,其余荧光碳量子点-纳米硒zeta电位值均大于30mv,均具有高的静电稳定性;而尿素和硼酸荧光碳量子点-纳米硒的荧光强度值和类氧化酶活性吸光值大,具有高的发光强度和类氧化酶活性。
84.实施例12:一种利用荧光碳量子点制备纳米硒的方法。
85.与实施例9相比,区别点仅在于:
86.以柠檬酸为碳源,以尿素为氮原子掺杂剂,分别以β-环糊精为表面钝化剂、硼酸为硼原子掺杂剂,在100℃常压水浴加热4h,合成共掺杂荧光碳量子点,以其为功能化软模板制备荧光碳量子点-纳米硒,其dh、总光强、pdi、电位、荧光强度和类氧化酶活性结果如图10所示。当以sds为表面钝化剂时,荧光碳量子点-纳米硒的dh较小、粒子数目较少、粒子粒径分布集中,zeta电位值大于30mv,类氧化酶活性吸光值最大,具有高的静电稳定性和类氧化酶活性。当以β-环糊精为表面钝化剂时,荧光碳量子点-纳米硒的dh小、粒子数目多、粒子粒径分布集中,zeta电位值和荧光强度值最大,具有高的静电稳定性和发光强度,但类氧化酶活性较低。
87.实施例13:一种利用荧光碳量子点制备纳米硒的方法。
88.与实施例12相比,区别点仅在于:
89.以抗坏血酸为碳源,分别以β-环糊精和sds为表面钝化剂,通过微波和水浴加热两种方式,合成共掺杂荧光碳量子点,以其为功能化软模板制备荧光碳量子点-纳米硒,其dh、总光强、pdi、电位、荧光强度和类氧化酶活性结果如图11所示。微波和水浴加热两种方式合成的荧光碳量子点均可有效稳定、分散纳米硒,并赋予其良好的荧光性能;其中以尿素为氮原子掺杂剂、sds为表面钝化剂时,水浴加热合成的荧光碳量子点制备的纳米硒的dh最小、静电稳定性最大,但以尿素为氮原子掺杂剂、β-环糊精为表面钝化剂时,微波加热合成的荧光碳量子点制备的纳米硒的类氧化酶活性最强。
90.实施例14:荧光碳量子点-纳米硒的胶体化学特性、荧光强度和类氧化酶活性比较。
91.综合比较实施例6、实施例9、实施例10、实施例11、实施例12、实施例13制备所得荧
光碳量子点-纳米硒的dh、原始光强、pdi、zeta电位、荧光强度、类氧化酶活性,结果如表1所示:
92.表1 荧光碳量子点-纳米硒的胶体化学特性、荧光强度和类氧化酶活性的绝对值吸光值比值
[0093][0094]
备注:sen范围值为1-8,值越大,荧光强度越弱。
[0095]
由表1结果可知,以柠檬酸为碳源、β-环糊精为表面钝化剂、尿素为氮原子掺杂化合物,经微波辅助加热合成的碳量子点制备的纳米硒dh最小、碳点稳定性和荧光强度最大,在hg
2+
作用下,具有最强的类氧化酶活性。
[0096]
实施例15:荧光碳量子点-纳米硒荧光探针检测hg
2+
。
[0097]
将0.2ml实施例6制备的荧光碳量子点-纳米硒与4.55ml的naac缓冲液(0.2mol/l,ph 5.0)混合,加入0.25ml不同浓度的hgcl2溶液,使其终浓度为0、0.7813、1.5625、3.1250、6.2500、12.5000、25.0000、50.0000、100.0000、200.0000、400.0000、800.0000μmol/l,25℃静置反应10min,于狭缝宽度为10nm、灵敏度为5、激发波长为420nm、扫描范围为350~550nm处测定其荧光光谱和荧光强度值,获得hg
2+
与荧光强度值的相关关系,建立hg
2+
检测方法,计算得到检出限(lod)。结果如图12所示:hg
2+
浓度(x)与荧光强度(y)具有较好的线性关系,经拟合得到线性曲线:y=-0.01217x+1.01964,r2=0.9931。lod为0.14μmol/l
[0098]
实施例16:荧光碳量子点-纳米硒荧光探针对hg
2+
的选择性。
[0099]
将0.2ml实施例6制备的荧光碳量子点-纳米硒与4.625ml的naac缓冲液(0.2mol/l,ph 5.0)混合,加入4mmol/l的na
+
、k
+
、ca
2+
、mg
2+
、mn
2+
、cu
2+
、zn
2+
、fe
2+
、ba
2+
、sr
2+
、co
2+
、cd
2+
、ni
2+
、pb
2+
、hg
2+
、cr
3+
、fe
3+
、al
3+
金属离子溶液(0.125ml,终浓度为0.1mmol/l),25℃静置反应10min,按实施例14参数测定其荧光光谱和荧光强度值。结果如图13所示:hg
2+
溶液可有效淬灭荧光碳量子点-纳米硒的荧光强度,为空白对照组的75%,其他金属离子溶液对荧光碳量子点-纳米硒的荧光强度无显著影响,荧光强度值均大于空白对照组95%以上,表明上述荧光碳量子点-纳米硒对hg
2+
具有高选择性。
[0100]
实施例17:基于hg
2+
触发荧光碳量子点-纳米硒的类氧化酶活性比色检测hg
2+
。
[0101]
将50μl实施例6制备的荧光碳量子点-纳米硒溶液(0.15mg/ml)与412.5μl的naac(0.2mol/l,ph 5.0)缓冲液混合,加入12.5μl不同浓度的hgcl2溶液,使其终浓度分别为18.75、25.00、31.25、37.50、43.75、50.00、75.00、100.00nmol/l,静置反应5min,加入5g/l显色底物tmb溶液25μl。反应体系置于30℃恒温水浴中,静置反应20min,取出置于冰浴中
30s,吸取200μl上述混合液于96孔板中,测定其在652nm处的吸光值a。测定空白组吸光值计a0。结果如图14所示:hg
2+
浓度(x)与类氧化酶活性的吸光绝对值比值(y,y=|a|/|a0|)呈现一定的线性关系,经拟合得到线性曲线:y=0.20081x-0.22672,r2=0.9823。lod为6.13nmol/l,低于世界卫生组织和美国环境保护署允许的饮用水中含hg
2+
的最高水平限值(30nmol/l和10nmol/l),满足检测要求。
[0102]
实施例18:荧光碳量子点-纳米硒的类氧化酶活性对hg
2+
的选择性。
[0103]
将50μl实施例6制备的荧光碳量子点-纳米硒悬液(0.15mg/ml)与412.5μl的naac(0.2mol/l,ph 5.0)缓冲液混合,分别加入12.5μl的na
+
、k
+
、ca
2+
、mg
2+
、mn
2+
、cu
2+
、zn
2+
、fe
2+
、ba
2+
、sr
2+
、ni
2+
、co
2+
、pb
2+
、cd
2+
、cr
3+
、al
3+
、fe
3+
金属离子溶液(2mmol/l和20mmol/l)或hgcl2溶液(2mmol/l),静置反应5min,加入5g/l显色底物tmb溶液25μl。反应体系置于30℃恒温水浴种,静置反应20min,取出置于冰浴中30s,吸取200μl上述混合液于96孔板中,测定其在652nm处的吸光值。结果如图15所示:hg
2+
可有效触发荧光碳量子点-纳米硒的类氧化酶活性,其他17种金属离子均不会改变荧光碳量子点-纳米硒的吸光绝对值比值,荧光碳量子点-纳米硒对hg
2+
具有高度特异性。
[0104]
实施例19:荧光碳量子点-纳米硒荧光-比色双信号检测hg
2+
的作用机制。
[0105]
将500μl实施例6制备的荧光碳量子点-纳米硒悬液(0.15mg/ml)与4.375ml的naac(0.2mol/l,ph 5.0)缓冲液混合,加入2mmol/l的hgcl2溶液125μl,反应体系置于30℃恒温水浴中,静置反应10min,取出置于冰浴中30s,溶液多次重复滴在锡箔纸上,测定xps图谱,分析c1s、se 3d和hg 4f高分辨图谱。结果如图16所示。hg
2+
作用荧光碳量子点-纳米硒后,在531.32、285.15、104.27、53.35ev处分别出现o1s、c 1s、hg 4f和se 3d的xps特征峰。se 3d分峰拟合图谱中电子结合能54.20ev与se在hgse的结合能非常吻合。hg 4f的电子结合能99.36、99.79、103.68ev与hg在hgse的结合能非常吻合,而104.24ev与hg
2+
的电子结合能非常吻合。基于上述结果初步推断,hg
2+
通过与荧光纳米硒配位,进而淬灭其荧光,同时与荧光碳量子点-纳米硒形成hgse,触发其类氧化酶活性。
[0106]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1