一种多孔碳材料快速石墨化的方法及其应用
1.本发明涉及新能源材料技术领域,具体涉及一种多孔碳材料快速石墨化的方法及其应用。
背景技术:
2.质子交换膜燃料电池(pemfc)能量转换的效率高,零污染排放(产物为水),这些优异的特点使得其成为21世纪尤为重要的新能源技术之一。由于大多数燃料电池所使用的催化剂为铂及铂基合金催化剂,为了使其能够充分被利用,常用碳材料作为载体,用于催化剂的分散。然而,由于碳载体在燃料电池实际工作中启停和长时间运行时,会产生严重的碳腐蚀,使负载在碳载体上的铂及铂基合金颗粒发生团聚,电化学活性面积降低,造成燃料电池的性能下降,且其所负载的催化剂性能越好,碳腐蚀越严重。使用石墨化的碳载体可以有效的缓解燃料电池在启停过程中所造成的碳腐蚀现象。然而我们通常制备的碳多数为硬碳,需要提升其石墨化程度。高温煅烧是最常用的石墨化的方法,然而往往需要高达2300℃的温度才能高度石墨化,而且需要升温降温的过程,耗时比较久,如中国专利202111243337.1。长时间的煅烧使原有的碳材料孔道减少,这将不利于催化剂的分散,因此,我们提出了一种高效快速提高碳载体石墨化缓解其碳腐蚀的方法,该方法操作简单,且反应快速,能够极大的缩短制备时间,防止多孔碳材料的骨架过多的分解形貌坍塌,且有效地保留了其高的比表面积,低温即可实现石墨化,该方法制备的碳材料将其应用在燃料电池催化剂载体上,可以有效的分散催化剂且有效的缓解碳腐蚀。
技术实现要素:
3.本发明的目的是为了快速高效地提高多孔碳载体的石墨化程度,提高其抗碳腐蚀的性能。同时提高多孔碳载体的石墨化程度的同时保留其多孔结构,使其可以利用碳载体的多孔结构在高温还原时限域催化剂,防止其团聚,有利于催化剂的分散。
4.为了克服现有的技术需要高于2000℃以上高温且升温降温所需要的时间较长,多孔结构容易坍塌的不足和缺点,本发明的目的在于提供一种多孔碳材料快速石墨化的方法及其应用(缓解燃料电池碳腐蚀)。
5.本发明的目的通过以下的技术方案实现。
6.一种多孔碳材料快速石墨化的方法,包括以下步骤:
7.(1)将石墨催化剂溶液和多孔碳材料混合,超声(石墨催化剂充分吸附在碳材料内)后干燥处理;
8.(2)将步骤(1)所得干燥处理后的混合材料进行超快速焦耳加热;
9.(3)将步骤(2)超快速焦耳加热后的混合材料进行酸洗(刻蚀反应产生的金属颗粒),得到石墨化的多孔碳材料。
10.优选的,步骤(1)中,所述石墨化催化剂溶液的溶剂为水或有机溶剂;进一步优选的,所述有机溶剂为乙醇或异丙醇。
11.优选的,步骤(1)所述石墨化催化剂为金属盐或硼酸;进一步优选的,所述金属盐为氯化钴,氯化锰,氯化铁,氯化镍,氯化铜,氯化锌,硝酸铁,硝酸钴,硝酸镍,硝酸锰,硝酸铜,醋酸铁,醋酸镍,乙酰丙酮钴和乙酰丙酮铁中的至少一种;
12.优选的,步骤(1)所述石墨化催化剂溶液的浓度为0.3-5g/ml。
13.优选的,步骤(1)所述的石墨化催化剂与多孔碳材料的质量比为(0.5-10)∶1。
14.优选的,步骤(1)所述超声的时间为0.5-24h,超声的功率为50-200w。所述干燥处理为冷冻干燥或鼓风干燥或红外灯烘干。冷冻干燥所需要冷井的温度为零下60℃-零下40℃,冷冻时间为2-6h,抽真空的时间为8-24h。鼓风干燥所需要的温度为40-80℃,所需时间为12-24h。红外灯的功率为50-100w,烘干所需时间为12-24h。
15.优选的,步骤(1)所述的多孔碳材料为多孔碳中的至少一种。
16.进一步优选的,步骤(1)所述多孔碳材料可为商业的炭黑如xc-72、bp2000、科琴黑等,也可为中空碳球、碗状碳、多孔硬碳、生物质衍生碳、mof衍生碳中多孔碳的至少一种;
17.优选的,步骤(1)所述多孔碳材料为块状或粉末状;所述多孔碳材料为粉末状,含有石墨催化剂和多孔碳材料的混合液干燥处理后加入到支撑载体装置中进行超快速焦耳加热。其中支撑载体装置材质可为碳纸、碳布、石墨纸等。
18.优选的,步骤(2)中,所述超快速焦耳加热的电压为10-40v,电流为10a-60a,加热的时间为20s-10min;所述超快速焦耳加热在惰性气体气氛中进行。
19.进一步优选的,步骤(2)中,所述超快速焦耳加热的电压为20-40v,电流为20a-50a,加热的时间为20s-100s;所述惰性气体为氩气。
20.优选的,步骤(3)中,所述酸洗的酸为稀硫酸、稀硝酸、稀盐酸和稀醋酸中的至少一种,浓度为0.3-4mol/l;所述酸洗的时间为6-24h,酸洗的温度可为30℃-80℃之间。
21.上述的方法得到的石墨化的碳材料在制备燃料电池催化剂中的应用。
22.优选的,包括以下步骤:将所述石墨化的碳材料浸渍氯铂酸和非贵金属盐溶液(其中氯铂酸的浓度为0.193mol/l),通过冷冻干燥和高温氢氩混合气退火还原,得到燃料电池碳载铂基催化剂。高温退火的温度为500-900℃,时间为2-12h;所述氢氩混合气中氢气的体积分数为5-20%。
23.进一步优选的,所述的非贵金属盐为硝酸钴,氯化钴,硫酸钴,醋酸钴,乙酰丙酮钴,硝酸镍,硝酸铁,硝酸锌,氯化锌,氯化铁,氯化锰和氯化铬中的至少一种。
24.与现有技术相比,本发明具有如下的有益效果:
25.本发明采用简单快速高效的焦耳加热方法制备得到高度石墨化的多孔碳载体,将石墨化催化剂和多孔碳等碳材料充分浸渍,干燥后通过超快速焦耳加热,再酸洗去除金属颗粒,得到高度石墨化的碳载体。此方法可以快速制备高度石墨化的碳载体,不需要长时间的高温煅烧,有效的缩短了反应时间,提高制备效率,且快速升温有利于碳材料孔结构的保留,使其可以利用碳载体的多孔结构在高温还原时限域催化剂,防止其团聚,有利于催化剂的分散。通过过渡金属的催化作用,低温即可实现石墨化,有利于节约能源。
附图说明
26.图1为实施例1的焦耳加热支撑载体装置示意图。
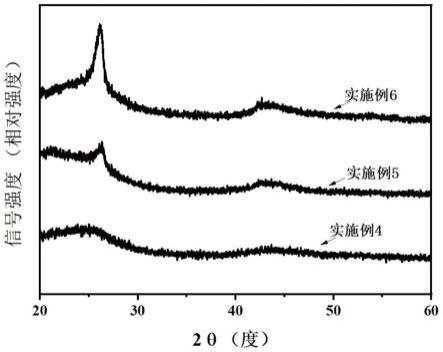
27.图2为实施例4-6制备的不同石墨化程度的多孔硬碳和未石墨化的多孔硬碳的xrd
对比图。
28.图3为实施例4-6制备的不同石墨化程度的多孔硬碳和未石墨化的多孔硬碳的拉曼对比图。
29.图4为实施例7制备的石墨化的硬碳和未处理硬碳负载金属间化合物pt3co的xrd对比图。
30.图5为实施例7制备的石墨化的硬碳、未处理硬碳负载金属间化合物pt3co和jm pt/c的高电位加速老化测试前后的氧化还原性能对比图。
具体实施方式
31.下面结合具体实施例对本发明作进一步地具体详细描述,但本发明的实施方式不限于此,对于为特别注明的工艺参数,可参照常规技术进行。
32.实施例1
33.(1)将1g六水合氯化铁加入到3ml水中,充分溶解后,加入500mgxc-72,充分浸渍超声10h后,冷冻干燥;
34.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中,如图1。设置恒电流为20a,电压为40v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
35.(3)将步骤(2)焦耳加热后的样品从支撑装置中取出,后浸入2m稀盐酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得高度石墨化的xc-72。
36.实施例2
37.(1)将1g醋酸铁加入到3ml水中,充分溶解后,加入500mgbp2000,充分浸渍超声10h后,冷冻干燥;
38.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中。设置恒电流为20a,电压为40v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
39.(3)将步骤(2)焦耳加热后的样品从碳支撑体中超声下来,后浸入0.5m稀盐酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得高度石墨化的bp2000。
40.实施例3
41.(1)将1g四水合氯化锰加入到3ml水中,充分溶解后,加入1g科琴黑,充分浸渍超声10h后,冷冻干燥;
42.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中。设置恒电流为20a,电压为40v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
43.(3)将步骤(2)焦耳加热后的样品从碳支撑体中超声下来,后浸入0.5m稀盐酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得高度石墨化的科琴黑。
44.实施例4
45.(1)将100mg多孔硬碳材料,放入超快加热支撑载体装置中。设置恒电流为10a,电压为10v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
46.(2)将步骤(1)焦耳加热后的样品浸入0.5m稀硫酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得处理后的硬碳。
47.实施例5
48.(1)将50mg六水合氯化铁加入到3ml水中,充分溶解后,加入100mg多孔硬碳,充分浸渍超声10h后,冷冻干燥;
49.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中。设置恒电流为10a,电压为10v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
50.(3)将步骤(2)焦耳加热后的样品浸入0.5m稀硫酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得处理后的硬碳。
51.实施例6
52.(1)将100mg六水合氯化铁加入到3ml水中,充分溶解后,加入100mg多孔硬碳,充分浸渍超声10h后,冷冻干燥;
53.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中。设置恒电流为20a,电压为40v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
54.(3)将步骤(2)焦耳加热后的样品浸入0.5m稀硫酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得高度石墨化的多孔硬碳。
55.图2为实施例4、实施例5、实施例6制备的不同石墨化程度的硬碳和未石墨化的硬碳的xrd对比图。图3为实施例4、实施例5、实施例6制备的不同石墨化程度的多孔硬碳和未石墨化的多孔硬碳的拉曼对比图。
56.从图2中可以看出未处理的硬碳(002)晶面的衍射角为25度左右,为比较宽的大鼓包衍射峰,说明其为无定形碳,经过石墨化后的硬碳,在26.2度附近出现很强的衍射峰,说明材料已经发生了明显的石墨化。同时从图3的拉曼图也可以看出,实施例6经过石墨化后的硬碳id/ig=0.6,说明碳材料的有序度增加,石墨化程度增强。而未石墨化的硬碳id/ig=1.03,缺陷程度较高,石墨化程度较低。
57.实施例7
58.(1)将2g六水合氯化铁加入到3ml水中,充分溶解后,加入2g多孔硬碳,充分浸渍超声10h后,冷冻干燥;
59.(2)将步骤(1)所得的样品,放入超快加热支撑载体装置中。设置恒电流为20a,电压为40v,时间设置为50s,氩气氛围下进行超快速焦耳加热;
60.(3)将步骤(2)焦耳加热后的样品取出,浸入0.5m稀硫酸溶液中,酸洗10h,然后过滤,用去离子水洗涤至中性,然后60℃烘干,即可得高度石墨化的硬碳。
61.(4)将步骤(3)所得的样品浸渍氯铂酸和钴盐,通过冷冻干燥和高温退火(750℃、2h,5℃/min),得到负载金属间化合物pt3co的高度石墨化碳复合物,并将其用于燃料电池高电位耐腐蚀测试中。(未处理多孔碳负载金属间化合物pt3co的制备方法同步骤(4))。
62.图4为实施例7制备的石墨化的多孔碳和未处理多孔碳负载金属间化合物pt3co的xrd图。从xrd中可以看出,pt3co已成功合成,通过半峰宽和谢乐公式可以得到制备的pt3co颗粒很小,说明石墨化后的多孔碳依然保持优异的孔道结构,依然能够在高温还原过程中有效的分散催化剂,这有利于催化剂电化学活性面积的增加,氧化还原性能的提高。
63.图5为实施例7制备的石墨化前后的多孔碳(hard carbon)负载金属间化合物pt3co(其中pt3co含量为20%)和jm pt/c(jm pt/c来自美国johnson matthey公司,其中pt含量为20%)的高电位(1-1.5v)加速老化测试(adt)前后的线性扫描伏安法(lsv)性能对比
图。通过lsv性能对比图可以看出,石墨化前后的多孔碳负载金属间化合物pt3co的催化性能均优于jm pt/c,但未石墨化的多孔碳基催化剂经过一万圈高电位的加速老化测试氧化还原性能衰减了58mv,而石墨化后的pt3co催化剂的氧化还原性能只衰减了5mv,商业jm pt/c衰减的10mv,显示出了比商业铂碳优异的耐腐蚀性能。
64.对所公开的实施例的上述说明,使本领域专业技术人员能够实现或者使用本发明。以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内,在其他实施例中实现。因此,本发明将不会被限制于这些实施例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1