一种用于药物载体的纳米碳酸钙制备方法
1.本发明涉及一种用于药物载体的纳米碳酸钙制备方法,属于材料工程、药物载体领域;应用于药物载体等。
背景技术:
2.碳酸钙是一种常见的无机化合物,具有成本低廉、无毒、无刺激性等优点。纳米级的碳酸钙在保留了以上优点的同时具备小尺寸效应,表面效应和量子尺寸效应,常用作药用辅料和补钙剂。近年来将纳米碳酸钙用于药物递送领域的相关报道引起了研究者的关注,纳米碳酸钙的多孔结构及高孔隙率有助于药物的装载与缓释,在碱性、中性环境下性质稳定,酸性环境下溶蚀分解的特性有助于药物的选择性递送。碳酸钙具有球霰石、文石和方解石三种晶型,其中球霰石是不稳定的碳酸钙晶型,具有良好的生物相容性、无毒性、可降解性的同时还有较高的比表面积,有利于药物的负载,特别适用于药物载体。
3.传统的碳化法在制备碳酸钙时,需要对反应温度、反应釜的压力以及通入的二氧化碳气体速率等条件进行控制才能制备得到纳米级的碳酸钙,其中晶型以方解石型,形貌以菱方形为主。碳化法的工艺繁琐、成本昂贵、操作复杂,无法实现对碳酸钙形貌与晶型的可控调节。复分解法利用水溶性钙盐和水溶性的碳酸盐在溶液中进行反应,在没有调控剂的情况下,生成的碳酸钙总是方解石型且粒径较大;当调控剂存在时,调控剂能够与钙离子或碳酸根粒子耦合,在反应体系中首先生成无定形碳酸钙颗粒,无定形碳酸钙颗粒在向其他晶型转变时,调控剂会影响晶胞的形成,从而诱导生成不同晶型与形貌的碳酸钙。聚丙烯酸是合成碳酸钙时常用的调控剂之一,聚天冬氨酸钠是工业上常用的阻垢剂,通过与钙离子交联耦合来形成无定形碳酸钙以抑制结垢,目前以聚天冬氨酸钠为晶型调控剂合成碳酸钙的研究中,只有少量关于无定形状态的碳酸钙的相关报道。本发明旨在解决上述问题,研发出一种可用于药物载体的纳米碳酸钙制备及晶型与形貌调控方法。
技术实现要素:
4.本发明的目的是解决制备纳米碳酸钙中存在的形貌与晶型不可控、制备工艺复杂的问题,提供一种用于药物载体的纳米碳酸钙制备方法,具有操作简单、成本低廉、晶型与形貌可控的优点。制备过程中无需复杂的设备,在烧杯中即可反应;制备成本低廉,晶型调控剂聚丙烯酸与聚天冬氨酸钠的价格便宜;得到的纳米碳酸钙由球霰石和方解石组成,其中球霰石的含量分布在30%~80%之间;纳米碳酸钙的形貌包括球状、棒状、球状与棒状的混合,随着球霰石含量的上升,碳酸钙的形貌存在从棒状向球状转变的趋势。制备得到的纳米碳酸钙均可作为药物载体,通过一个简单的制备方法可以得到纳米碳酸钙-药物复合制剂。
5.调控剂聚天冬氨酸钠和聚丙烯酸的使用,使得调控剂与钙离子或碳酸钠离子形成耦合物,能够在反应体系中形成较稳定的形成无定形态的碳酸钙颗粒。无定形态的碳酸钙能够在反应体系中稳定存在,但在离心过程中,无定形态的碳酸钙会转变为其他晶型。调控
剂会影响晶胞的形成,诱导形成粒径更小具有不同形貌和晶型的碳酸钙。
6.本发明制备纳米碳酸钙的化学方程式如下:
7.cacl2+na2co3=ca2co3↓
+2nacl
8.本发明解决以上问题所采用的技术方案如下:一种用于药物载体的纳米碳酸钙制备方法步骤如下:
9.步骤一、将氯化钙溶解于1mm、ph为7.6的tris-hcl缓冲溶液中,氯化钙浓度为0.1mol/l;将碳酸钠溶解于1mm、ph为7.6的hepes缓冲溶液中,碳酸钠浓度为0.1mol/l;
10.步骤二、将调控剂聚天冬氨酸钠/聚丙烯酸加入氯化钙/碳酸钠溶液中,搅拌交联30min,制得氯化钙-聚天冬氨酸钠/聚丙烯酸-碳酸钠混合溶液;聚天冬氨酸钠与氯化钙的质量比为3∶1,聚丙烯酸与碳酸钠的质量比为0.25~0.5∶1;
11.步骤三、将碳酸钠溶液以10ml/min的速率滴加至氯化钙溶液中,碳酸钠与氯化钙的质量比为106∶111,室温下搅拌12~24h;
12.步骤四、反应结束后,通过12000r/min离心反应液30min,分离得到固体,用10ml超纯水、10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
13.步骤五、干燥后得到的纳米碳酸钙,由球霰石和方解石组成,其中球霰石的含量为30~80%,方解石的含量为20~70%;纳米碳酸钙的形貌为球状、棒状、球状、棒状中的一种以上。
14.采用聚天冬氨酸钠作为调控剂制备球状形貌纳米碳酸钙,制备方法步骤如下:
15.步骤一、将氯化钙溶解于1mm、ph为7.6的tris-hcl缓冲溶液中,氯化钙浓度为0.1mol/l;将碳酸钠溶解于1mm、ph为7.6的hepes缓冲溶液中,碳酸钠浓度为0.1mol/l;
16.步骤二、将调控剂聚天冬氨酸钠加入氯化钙溶液中,聚天冬氨酸钠与氯化钙质量比为3∶1,搅拌交联30min;
17.步骤三、将碳酸钠溶液以10ml/min的速率滴加至氯化钙-聚天冬氨酸钠混合溶液中,碳酸钠与氯化钙的质量比为106∶111,室温下搅拌12~24h;
18.步骤四、反应结束后,通过12000r/min离心反应液30min,分离得到固体,用10ml超纯水、10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
19.步骤五、经干燥后得到的球状形貌纳米碳酸钙由77%的球霰石、23%的方解石组成。
20.采用聚丙烯酸作为调控剂制备棒状形貌纳米碳酸钙,制备方法步骤如下:
21.步骤一、将氯化钙溶解于1mm、ph为7.6的tris-hcl缓冲溶液中,氯化钙浓度为0.1mol/l;将碳酸钠溶解于1mm、ph为7.6的hepes缓冲溶液中,碳酸钠浓度为0.1mol/l;
22.步骤二、将调控剂聚丙烯酸加入碳酸钠溶液中,聚丙烯酸与碳酸钠质量比为0.25∶1,搅拌交联30min;
23.步骤三、将碳酸钠-聚丙烯酸混合溶液以10ml/min的速率滴加至氯化钙溶液中,碳酸钠与氯化钙的质量比为106∶111,室温下搅拌12~24h;
24.步骤四、反应结束后,通过12000r/min离心反应液30min,分离得到固体,用10ml超纯水、10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
25.步骤五、经干燥后得到的棒状形貌纳米碳酸钙由36%的球霰石和64%的方解石组成。
26.采用聚天冬氨酸钠和聚丙烯酸作为调控剂制备球状形貌与棒状形貌混合纳米碳酸钙,制备方法步骤如下:
27.步骤一、将氯化钙溶解于1mm、ph为7.6的tris-hcl缓冲溶液中,氯化钙浓度为0.1mol/l;将碳酸钠溶解于1mm、ph为7.6的hepes缓冲溶液中,碳酸钠浓度为0.1mol/l;
28.步骤二、将调控剂聚天冬氨酸钠加入氯化钙溶液中,聚天冬氨酸钠与氯化钙质量比为3∶1;将调控剂聚丙烯酸加入碳酸钠溶液中,聚丙烯酸与碳酸钠质量比为0.5∶1;搅拌交联30min;
29.步骤三、将碳酸钠-聚丙烯酸混和溶液以10ml/min的速率滴加至氯化钙-聚天冬氨酸钠混合溶液中,碳酸钠与氯化钙的质量比为106∶111,室温下搅拌12~24h;
30.步骤四、反应结束后,通过12000r/min离心反应液30min,分离得到固体,用10ml超纯水、10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
31.步骤五、经干燥后得到的球状形貌与棒状形貌混合纳米碳酸钙晶型由57%的球霰石型、43%的方解石型组成。
32.纳米碳酸钙-药物复合制剂的方法步骤如下:
33.将纳米碳酸钙载体和药物按照质量比为1∶2同时分散至20ml水中,45khz频率下超声30min、磁力搅拌4h混合均匀后,过滤收集固态复合制剂,用10ml水洗涤后冷冻干燥,制得纳米碳酸钙-药物复合制剂。
34.制备得到的复合制剂具有缓释作用,持续释药时间大于48h。
35.装载药物为奥曲肽、枸橼酸喷托维林或吡嗪酰胺。
36.本发明的有益效果:(1)制备的纳米碳酸钙,能够对产品的晶型及形貌进行可控调节的同时具有操作简单、成本低廉的优点,制备得到的纳米碳酸钙均可作为药物载体。
37.(2)纳米碳酸钙-药物复合制剂制备方法能够简单高效的装载药物。
38.(3)纳米碳酸钙能够有效装载大分子多肽药物和小分子药物,制备的纳米碳酸钙-药物复合制剂具有缓释作用,用于干粉吸入制剂与纳米混悬注射制剂。
附图说明
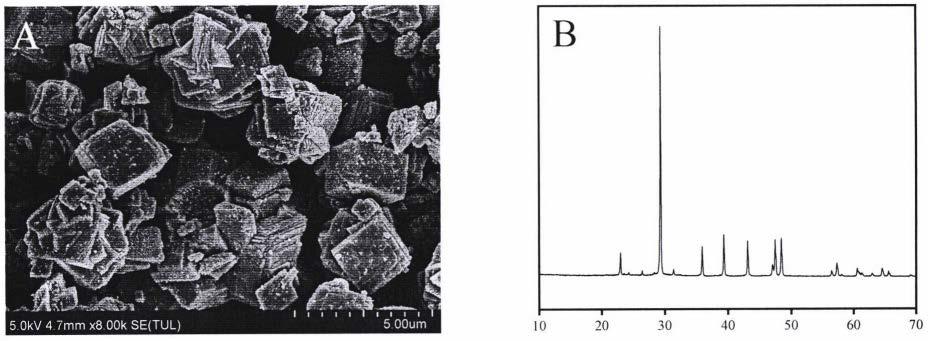
39.图1菱方状形貌微米碳酸钙的扫描电镜图及xrd图
40.图中:a为扫描电镜图,图b为xrd图。
41.图2菱方状形貌微米碳酸钙载药前后的透射电镜图
42.图中:图a为载药前的透射电镜图,图b为载药后的透射电镜图。
43.图3菱方状形貌微米碳酸钙-奥曲肽复合制剂的体外溶出曲线
44.图中:溶出介质为37℃,ph为7.4的pbs缓冲溶液;横坐标为溶出数据,单位为min;纵坐标为溶出度,单位为%;线a代表菱方状形貌微米碳酸钙-奥曲肽复合制剂,线b代表纯奥曲肽。
45.图4球状形貌纳米碳酸钙的扫描电镜图及xrd图
46.图中:图a为扫描电镜图,图b为xrd图。
47.图5球状形貌纳米碳酸钙载药前后的透射电镜图
48.图中:图a为载药前的透射电镜图,图b为载药后的透射电镜图。
49.图6球状形貌纳米碳酸钙-奥曲肽复合制剂的体外溶出曲线
50.图中:溶出介质为37℃,ph为7.4的pbs缓冲溶液;横坐标为溶出数据,单位为min;纵坐标为溶出度,单位为%;图中线a代表球状形貌纳米碳酸钙-奥曲肽复合制剂,线b代表纯奥曲肽。
51.图7棒状形貌纳米碳酸钙的扫描电镜图及xrd图
52.图中:图a为扫描电镜图,图b为xrd图。
53.图8球状形貌与棒状形貌混合纳米碳酸钙的扫描电镜图及xrd图
54.图中:图a为扫描电镜图,图b为xrd图。
具体实施方式
55.下面结合实施例和附图对本发明作进一步说明。
56.实施例1(对比实施例)
57.菱方状形貌微米碳酸钙的制备及菱方状形貌微米碳酸钙-奥曲肽复合制剂的制备
58.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将碳酸钠溶液以10ml/min的速率滴加至氯化钙溶液中,室温下以1000r/min的转速搅拌12h;反应结束后,将反应液在12000r/min的转速下离心30min得到菱方状形貌微米碳酸钙,用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;制备的菱方状形貌微米碳酸钙为方解石型,其扫描电镜图及xrd图如图1所示。
59.将50mg菱方状形貌微米碳酸钙载体和100mg的奥曲肽同时分散至20ml水中,在45khz频率下超声30min、磁力搅拌4h;菱方状形貌微米碳酸钙充分吸附药物后,过滤收集固态复合制剂,用少量水洗涤后冷冻干燥。菱方状形貌微米碳酸钙载药前后的透射电镜图如图2所示,药物附着在载体的表面,载药量为3.35%,有效部位沉积率为34%。
60.对菱方状形貌微米碳酸钙-奥曲肽复合制剂进行体外溶出性能的测定。以37℃,ph=7.4的pbs缓冲溶液为溶出介质;在溶出开始后的第1mm、10min、30min、60min、240min、720min、1440min、2880min取样并进行定量分析,然后补充等体积等温的pbs缓冲溶液;溶出曲线如图3所示。与纯oct相比,菱方状形貌微米碳酸钙-奥曲肽复合制剂迅速溶出,不具备缓释作用。
61.实施例2
62.球状形貌纳米碳酸钙的制备及球状形貌纳米碳酸钙-奥曲肽复合制剂的制备
63.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将999mg的聚天冬氨酸钠加入氯化钙溶液中搅拌交联30min;将碳酸钠溶液以10ml/min的速率滴加至氯化钙-聚天冬氨酸钠混合溶液中,室温下以1000r/min的转速搅拌12h;反应结束后,将反应液在12000r/min的转速下离心30min得到球状形貌纳米碳酸钙;用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;制备的球状形貌纳米碳酸钙中含有77%的球霰石和23%的方解石,其扫描电镜与xrd图如图4所示。
64.将50mg球状形貌纳米碳酸钙和100mg的奥曲肽同时分散至20ml水中,在45khz频率下超声30min、磁力搅拌4h;球状形貌纳米碳酸钙充分吸附药物后,过滤收集固态复合制剂,用少量水洗涤后冷冻干燥。球状形貌纳米碳酸钙载药前后的透射电镜图如图5所示,球状形
貌纳米碳酸钙载药后的颜色明显加深,药物被装载了它内部的孔道之中,载药量为20.3%,有效部位沉积率为41.4%。
65.对球状形貌纳米碳酸钙-奥曲肽复合制剂进行体外溶出性能的测定。以37℃,ph=7.4的pbs缓冲溶液为溶出介质;在溶出开始后的第1min、10min、30min、60min、240min、720min、1440min、2880min取样并进行定量分析,然后补充等体积等温的pbs缓冲溶液;溶出曲线如图6所示。与纯oct相比,球状形貌纳米碳酸钙-奥曲肽复合制剂持续释药48h,具备较好的缓释作用。
66.实施例3
67.棒状形貌纳米碳酸钙的制备
68.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将79.5mg的聚丙烯酸加入碳酸钠溶液中,搅拌交联30min;将碳酸钠-聚丙烯酸混合溶液以10ml/min的速率滴加至氯化钙溶液中,室温下以1000r/min的转速搅拌12h;反应结束后,将反应液在12000r/min的转速下离心30min得到棒状形貌纳米碳酸钙;用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;制备的棒状形貌纳米碳酸钙晶型中含有36%的球霰石和64%的方解石,其扫描电镜图和xrd图如图7所示。
69.实施例4
70.球状形貌与棒状形貌混合纳米碳酸钙的制备
71.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将999mg的聚天冬氨酸钠加入氯化钙溶液中,将159mg的聚丙烯酸加入碳酸钠溶液中,搅拌交联30min;将碳酸钠-聚丙烯酸混和溶液以10ml/min的速率滴加至氯化钙-聚天冬氨酸钠混合溶液中,室温下以1000r/min的转速搅拌12h;反应结束后,将反应液在12000r/min的转速下离心30min,得到球状形貌与棒状形貌混合纳米碳酸钙;用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;制备的棒状形貌纳米碳酸钙晶型中含有57%的球霰石和43%的方解石,其扫描电镜图和xrd图如图8所示。
72.实施例5
73.球状形貌纳米碳酸钙的制备及球状形貌纳米碳酸钙-吡嗪酰胺复合制剂的制备
74.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将999mg的聚天冬氨酸钠加入氯化钙溶液中搅拌交联30min;将碳酸钠溶液以10ml/min的速率滴加至氯化钙-聚天冬氨酸钠混合溶液中;室温下以1000r/min的转速搅拌24h;反应结束后,将反应液在12000r/min的转速下离心30min得到球状形貌纳米碳酸钙;用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
75.将50mg球状形貌纳米碳酸钙和100mg的吡嗪酰胺同时分散至20ml水中,在45khz频率下超声30min、磁力搅拌4h;球状形貌纳米碳酸钙充分吸附药物后,过滤收集固态复合制剂,用少量水洗涤后冷冻干燥。载药量为26%,有效部位沉积率为62%。
76.实施例6
77.棒状形貌纳米碳酸钙的制备及球状形貌纳米碳酸钙-枸橼酸喷托维林复合制剂的
制备
78.将333mg氯化钙溶解于30ml的tris-hcl缓冲溶液(1mm,ph=7.6)中,氯化钙浓度为0.1mol/l;将318mg碳酸钠溶于30ml的hepes缓冲溶液(1mm,ph=7.6)中,碳酸钠浓度为0.1mol/l;将79.5mg的聚丙烯酸加入碳酸钠溶液中,搅拌交联30min;将碳酸钠-聚丙烯酸混合溶液以10ml/min的速率滴加至氯化钙溶液中,室温下以1000r/min的转速搅拌24h;反应结束后,将反应液在12000r/min的转速下离心30min得到棒状形貌纳米碳酸钙;用10ml超纯水和10ml乙醇交替洗涤,重复三次,50℃下干燥至恒重;
79.将50mg棒状形貌纳米碳酸钙和100mg的枸橼酸喷托维林同时分散至20ml水中,在45khz频率下超声30min、磁力搅拌4h;棒状形貌纳米碳酸钙充分吸附药物后,过滤收集固态复合制剂,用少量水洗涤后冷冻干燥。载药量为30%,有效部位沉积率为51%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1