由6,10‑二甲基十一碳‑5‑烯‑2‑酮或6,10‑二甲基十一碳‑5,9‑二烯‑2‑酮制备的(6R,10R)‑6,10,14‑三甲基十五烷‑2‑酮的制作方法与工艺
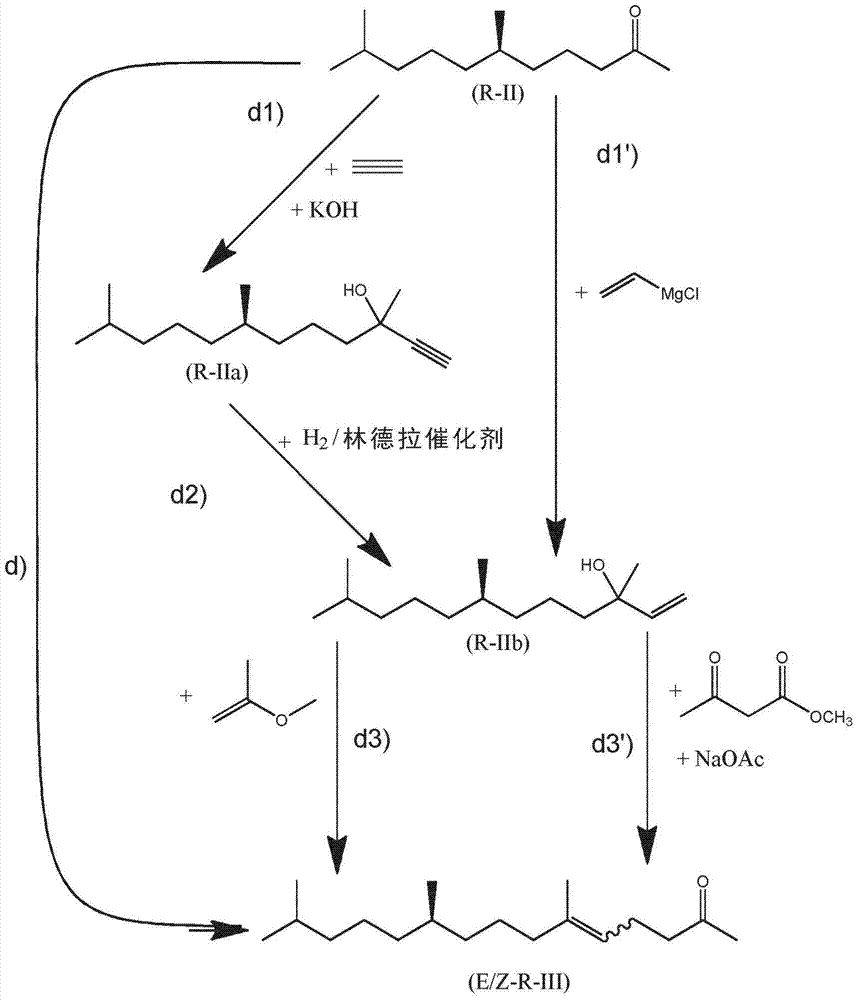
由6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮制备的(6R,10R)-6,10,14-三甲基十五烷-2-酮技术领域本发明涉及(6R,10R)-6,10,14-三甲基十五烷-2-酮及其反应产物的领域。发明背景(6R,10R)-6,10,14-三甲基十五烷-2-酮是一种重要的中间体,特别是对于合成(R,R)-异植醇[=(3RS,7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇]、(R,R)-植醇和生育酚而言。异植醇、植醇和生育酚是手性物质,其中后两种以“全R”立体异构体的形式存在于自然界中。植醇包含2个立构中心以及另外的产生E/Z-立体异构体的三取代的碳-碳双键,而异植醇和生育酚具有3个立构中心。因此,存在多种异构体。已证实,天然存在的生育酚的立体异构体(2R,4’R,8’R)-生育酚,特别是(2R,4’R,8’R)-α-生育酚,具有最高的生物活性(生物效能)。然而,由于(2R,4’R,8’R)-生育酚和(R,R)-植醇的天然来源非常有限,因此市场上对于(2R,4’R,8’R)-生育酚和(R,R)-异植醇以及(6R,10R)-6,10,14-三甲基十五烷-2-酮的有效合成有强烈的需求,(6R,10R)-6,10,14-三甲基十五烷-2-酮为这些产物的起始物料,其适用于工业规模应用。此外,由于例如H.Weiser等人在J.Nutr.1996,126(10),2539-49中已证实,在位于分子环中醚氧原子附近的手性中心处具有R-构型(即,2R-构型)的生育酚与具有S-构型的对应异构体相比表现出更高的生物活性(生物效能),因此对(2R,4’R,8’R)-生育酚,特别是(2R,4’R,8’R)-α-生育酚的有效工业规模合成有强烈的需求。
技术实现要素:
因此,本发明要解决的问题是提供一种制备(6R,10R)-6,10,14-三甲基十五烷-2-酮的方法。令人惊奇的是,已发现此问题可通过根据权利要求1所述的方法来解决。已证实,可由起始物料的异构体混合物获得一种感兴趣的特定异构体,所述混合物即为(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的混合物或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮和(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的混合物。本发明的优选实施方式允许通过使用顺式/反式异构化来利用非期望的异构体。不对称氢化是本发明的关键要素之一,其可通过将待被不对称氢化的酮缩酮化以及使用特定的添加剂来在质量和速度方面得到改善。本发明的方法允许由异构混合物高效且高质量地制备目标分子,从而允许其用于工业规模制备。所述方法是非常有利的,因为其以有效的方式由起始产物的立体异构体混合物形成期望的手性产物。本发明的另外的方面为另外的独立权利要求的主题。特别优选的实施方案是从属权利要求的主题。具体实施方式在第一方面,本发明涉及一种以多步合成的方式由6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮制备(6R,10R)-6,10,14-三甲基十五烷-2-酮的方法,其包括以下步骤:a)提供(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的混合物或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮和(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的混合物;b)将6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的一种异构体从步骤a)的混合物分离;c)在手性铱络合物的存在下使用分子氢进行不对称氢化并且生成(R)-6,10-二甲基十一烷-2-酮;d)将(R)-6,10-二甲基十一烷-2-酮化学转化成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物;e)将(R)-6,10,14-三甲基十五碳-5-烯-2-酮的一种异构体从步骤d)中获得的混合物分离;f)在手性铱络合物的存在下使用分子氢进行不对称氢化并且生成(6R,10R)-6,10,14-三甲基十五烷-2-酮;其中所述步骤a)-f)的顺序为a、b、c、d、e、f。在本文件中术语"彼此独立地"是指在取代基、部分或基团的上下文中,相同指定的取代基、部分或基团可在同一分子中同时以不同的含义出现。“Cx-y-烷基”基团是包含x至y个碳原子的烷基基团,即,例如C1-3-烷基基团是包含1至3个碳原子的烷基基团。烷基基团可为直链或支链。例如,-CH(CH3)-CH2-CH3被视为C4-烷基基团。“Cx-y-亚烷基”基团是包含x至y个碳原子的亚烷基基团,即,例如C2-C6亚烷基基团是包含2至6个碳原子的烷基基团。亚烷基基团可为直链或支链的。例如,基团-CH(CH3)-CH2-被视为C3-亚烷基基团。在本文件中“酚醇”是指羟基与芳族基团直接结合的醇。本文件中使用的术语“(R,R)-异植醇”是指(3RS,7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇)。本文件中使用的术语“(R,R)-植醇”是指(2E,7R,11R)-3,7,11,15-四甲基-2-十六碳烯-1-醇)。如本文件中使用时,以“聚”为首的物质名称如聚硫醇,是指在形式上每分子包含两个或更多个相应官能团的物质。如本文件中使用时,术语“立体异构中心”是携带多个如下基团的原子,这些基团中的任何两个基团之间的互换导致产生立体异构体。立体异构体是这样的异构分子,该异构分子具有相同分子式和键合原子的顺序(构成)但它们的原子在空间上的三维取向不同。立体异构中心处的构型被限定为R或S。确定立体化学中绝对构型的R/S概念和规则对于本领域的技术人员而言是已知的。在本文件中,如果将分子氢加至碳-碳双键导致形成立体异构碳中心,则所述碳-碳双键被定义为“前手性的”。顺式/反式异构体为在双键处具有不同取向的构型异构体。在本文件中,术语“顺式”的使用等同于“Z”(反之亦然),并且“反式”的使用等同于“E”(反之亦然)。因此,例如,术语“顺式/反式异构化催化剂”等同于术语“E/Z异构化催化剂”。“顺式/反式异构化催化剂”是能够将顺式异构体(Z-异构体)异构化成顺式/反式异构体混合物(E/Z异构体混合物)或将反式异构体(E-异构体)异构化成顺式/反式异构体(E/Z异构体混合物)的催化剂。术语“E/Z”、“顺式/反式”和“R/S”分别表示E和Z的混合物、顺式和反式的混合物以及R和S的混合物。如果符号或基团的相同标记存在于数个式中,则在本文件中,一个具体的式的语境中对所述基团或符号所做的定义也适用于包括所述相同标记的其他式。在本文件中,任何单虚线均表示取代基与分子的其余部分结合的键。不对称氢化的"测定收率"(assayyield)在本申请中是完全饱和的酮或醛或缩酮或缩醛的分子数与将经受氢化的不饱和酮或醛或缩酮或缩醛的分子数的摩尔比。6,10-二甲基十一碳-5-烯-2-酮是一种已知的物质,并且可例如由3,7-二甲基辛-1-烯-3-醇和2-甲氧基丙-1-烯合成,如DE1193490中所公开。3,7-二甲基辛-1-烯-3-醇可例如根据WO2012/025559A2中所公开的程序来被合成。DE1193490和WO2012/025559A2的全部内容通过引用结合于此。6,10-二甲基十一碳-5-烯-2-酮是(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的混合物。6,10-二甲基十一碳-5,9-二烯-2-酮可商购获得并且可例如通过卡莱尔重排(Carrollrearrangement)由里那醇合成,并且是(E)-6,10-二甲基十一碳-5,9-二烯-2-酮和(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的混合物。在所述方法的第一步骤(步骤a)中,提供(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的混合物或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮和(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的混合物。分离步骤b)和e)分别涉及将6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的异构体中的一种从步骤a)的混合物分离,以及将(R)-6,10,14-三甲基十五碳-5-烯-2-酮的异构体中的一种从步骤d)中获得的混合物分离。步骤b)和/或e)中的这种异构体分离可以不同的方式进行。第一种可能是通过色谱法来分离。另一种优选的分离方式是通过蒸馏来进行步骤b)和/或e)中的异构体的分离。所述分离由于所述异构体具有不同沸点而成为可能。为了最小化异构体的热降解,可取的是在减压下通过蒸馏柱来进行蒸馏。由于待分离的异构体具有不同的沸点(参见表1),因此异构体可通过蒸馏来分离。通过使用特定的蒸馏技术和设备,可分离具有更高或更低沸点的异构体。物质沸点(E)-6,10-二甲基十一碳-5-烯-2-酮5毫巴下112℃(Z)-6,10-二甲基十一碳-5-烯-2-酮5毫巴下109℃(E)-6,10-二甲基十一碳-5,9-二烯-2-酮0.5毫巴下78℃(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮0.5毫巴下75℃(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮2毫巴下122℃(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮2毫巴下119℃表1:异构体的沸点。在一个优选的实施方式中,蒸馏在顺式/反式异构化催化剂的存在下进行。顺式/反式异构化顺式/反式异构化催化剂为使碳-碳双键异构化的催化剂。已发现,就本发明的目的而言,催化5位和9位中双键的顺式/反式异构化的所述顺式/反式异构化催化剂具体为一氧化氮(NO)或有机硫化合物,具体为聚硫醇。特别适合作为顺式/反式异构化催化剂的是式(X)的聚硫醇或芳族聚硫醇其中n1表示1至4的整数,特别是2,并且m1表示2至8的整数,特别是3或4,优选4;并且A表示分子量介于28g/mol和400g/mol之间,特别是介于90g/mol和150g/mol之间的脂族m1-价烃基。聚硫醇季戊四醇四(巯基乙酸酯)、三羟甲基丙烷三(巯基乙酸酯)、二巯基乙酸乙二醇酯、季戊四醇四-(3-巯基丙酸酯)、三羟甲基丙烷三-(3-巯基丙酸酯)(=2-乙基-2-(((3-巯基丙酰基)氧基)甲基)丙烷-1,3-二基双(3-巯基丙酸酯),2-ethyl-2-(((3-mercaptopropanoyl)oxy)methyl)propane-1,3-diylbis(3-mercaptopropanoate))和乙二醇二-(3-巯基丙酸酯)已被证实是高度优选的式(X)的聚硫醇并且是所有上述聚硫醇中的优选聚硫醇。特别优选的作为芳族聚硫醇的是4,4'-二巯基联苯或4,4'-硫代二苯硫醇。将式(X)的聚硫醇用作顺式/反式异构化催化剂是非常有利的,因为聚硫醇通常具有非常低的蒸气压(即高沸点),从而使得其可在高温下使用,例如同时蒸馏低沸点异构体。此外,每分子量的聚硫醇具有高密度的硫醇官能团,这是非常有利的,因为仅需添加少量的催化剂。将聚硫醇用作顺式/反式异构化催化剂是非常有利的,因为其允许非常快速的异构化。一氧化氮(NO)是一种气体并且可原样或以气体混合物(特别是与至少一种惰性的气体组合,特别是与氮气组合)的形式被引入待异构化的酮或缩酮中。如果使用气体混合物,则气体混合物中的一氧化氮的量按所述气体混合物的重量计优选地在1%-99%,特别是5%-95%的范围内。具体而言,鉴于腐蚀和毒性,气体混合物中的一氧化氮的量按所述气体混合物的重量计优选地在10%-60%的范围内。将一氧化氮用作顺式/反式异构化催化剂是非常有利的,因为这种异构化催化剂可容易地从待异构化的酮或缩酮中移除。优选地将一氧化氮在大气压力或至多1MPa的过压下引入酮或缩酮。过压优选地等于10至300kPa。一氧化氮(NO)或一氧化氮(NO)与其它气体的混合物优选地通过管以连续的方式被引入,并且被鼓泡通过待异构化的酮或缩酮。顺式/反式异构化的使用允许使纯的顺式或反式异构体或这些异构体的任意混合物转化,生成顺式和反式异构体的热力学平衡的混合物。总之,这使得能够通过蒸馏来分离期望的异构体以及使得非优选异构体(残留异构体)能够转化(异构化)成期望的异构体。蒸馏可在顺式/反式异构化催化剂的存在下进行(一锅异构化或原位异构化),使得期望的异构体连续再形成并且可通过蒸馏来被分离。此外,顺式/反式异构化可在单独/分开的容器中进行,在所述单独/分开的容器中,顺式/反式异构化催化剂被添加至蒸馏残余物中。因此,残留异构体通过顺式/反式异构化催化剂而被异构化,并且随后被添加至分别在步骤a)和d)中提供的异构体的相应混合物中。在步骤b)和/或e)中使用顺式/反式异构化使得期望的异构体具有高收率。在优选的情况中,可实现基本上将所有的非期望异构体全部异构化成期望的异构体。优选地,特别是在异构化催化剂不是一氧化氮的情况中,更优选地在聚硫醇作为异构化催化剂的情况中,在高于20℃的温度下、特别是在介于20℃和期望的异构体的沸点之间的温度下、特别是在介于50℃和期望的异构体的沸点之间的温度下进行异构化。异构化可在环境压力下或在减压下进行。在一锅异构化的情况中,异构化优选地在减压下进行。特别是对于一氧化氮为顺式/反式异构化催化剂的情况,异构化在环境压力或过压下进行。已进一步观察到,在采用聚硫醇的异构化中,添加极性溶剂诸如酰胺、吡咯烷酮、砜、亚砜、离子液体,特别是N,N-二甲基甲酰胺(DMF)或N-甲基-2-吡咯烷酮(NMP)、环丁砜、二甲基亚砜(DMSO)和1-丁基-3-甲基咪唑鎓溴化物对异构化具有加速作用。因此,优选的是顺式/反式异构化的过程在极性溶剂,特别是在选自由离子液体,特别是1-丁基-3-甲基咪唑鎓溴化物、N,N-二甲基甲酰胺(DMF)、N-甲基-2-吡咯烷酮(NMP)、环丁砜和二甲基亚砜(DMSO)组成的极性溶剂的存在下进行。更优选的是顺式/反式异构化的过程在极性溶剂,特别是在选自由离子液体,特别是1-丁基-3-甲基咪唑鎓溴化物、N,N-二甲基甲酰胺(DMF)、N-甲基-2-吡咯烷酮(NMP)和二甲基亚砜(DMSO)组成的极性溶剂的存在下进行。分别相对于6,10-二甲基十一碳-5-烯-2-酮的异构体的量或6,10-二甲基十一碳-5,9-二烯-2-酮的异构体的量、相对于(R)-6,10,14-三甲基十五碳-5-烯-2-酮的量,顺式/反式异构化催化剂的量优选地介于1重量%和20重量%之间。缩酮形成在另一个实施方式中,在步骤c)之前进行步骤co)co)形成步骤b)中分离的6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的异构体的缩酮。因此,在步骤c)中,6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮被不对称氢化,并且在不对称氢化后,经氢化的缩酮被水解成酮并且生成(R)-6,10-二甲基十一烷-2-酮。在另一个实施方案中,在步骤f)之前进行步骤fo)fo)形成步骤e)中分离的(R)-6,10,14-三甲基十五碳-5-烯-2-酮的异构体的缩酮因此,在步骤f)中,(R)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮被不对称氢化,并且在不对称氢化后,经氢化的缩酮被水解成酮并且生成(6R,10R)-6,10,14-三甲基十五烷-2-酮。由酮本身形成缩酮对于本领域的技术人员而言是已知的。不饱和酮的缩酮可优选地由上述不饱和酮和醇形成。对于本领域的技术人员而言已知的是存在替代的缩酮合成路线。原则上,缩酮还可通过用原酸酯处理酮或通过反式缩酮化来形成,例如公开于如Pério等人的TetrahedronLetters1997,38(45),7867-7870或Lorette和Howard的J.Org.Chem.1960,521-525中,这两篇文献的全部内容通过引用结合于此。优选地,缩酮由上述不饱和酮和醇形成。醇可包含一个或多个羟基基团。醇可为酚醇或脂族或脂环族醇。优选地,醇具有一个或两个羟基基团。如果醇具有一个羟基基团,则醇优选地为具有1至12个碳原子的醇。具体而言,具有一个羟基基团的醇选自由如下组成的组:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-甲基-1-丙醇、2-丁醇、戊烷-1-醇、3-甲基丁烷-1-醇、2-甲基丁烷-1-醇、2,2-二甲基丙-1-醇、戊烷-3-醇、戊烷-2-醇、3-甲基丁烷-2-醇、2-甲基丁-2-醇、己烷-1-醇、己烷-2-醇、己烷-3-醇、2-甲基-1-戊醇、3-甲基-1-戊醇、4-甲基-1-戊醇、3-甲基-2-戊醇、4-甲基-2-戊醇、2-甲基-3-戊醇、2,2-二甲基-1-丁醇、2,3-二甲基-1-丁醇、3,3-二甲基-1-丁醇、3,3-二甲基-2-丁醇、2-乙基-1-丁醇,以及庚醇、辛醇和卤代C1-C8-烷基醇的全部结构异构体,特别是2,2,2-三氟乙醇。特别合适的是伯醇或仲醇。优选地,将伯醇用作具有一个羟基基团的醇。具体而言,将甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇或2,2,2-三氟乙醇,优选甲醇、乙醇、1-丙醇、1-丁醇或2,2,2-三氟乙醇用作具有一个羟基基团的醇。在另一个实施方案中,醇为二醇,因此具有两个羟基基团。优选地,二醇选自由如下组成的组:乙烷-1,2-二醇、丙烷-1,2-二醇、丙烷-1,3-二醇、丁烷-1,4-二醇、丁烷-1,3-二醇、丁烷-1,2-二醇、丁烷-2,3-二醇、2-甲基丙烷-1,2-二醇、2,2-二甲基丙烷-1,3-二醇、1,2-二甲基丙烷-1,3-二醇、苯-1,2-二醇和环己烷-1,2-二醇。就两种环己烷-1,2-二醇而言,优选的立体异构体为同式-环己烷-1,2-二醇(syn-cyclohexane-1,2-diol)(=顺式-环己烷-1,2-二醇)。在一个实施方式中,两个羟基基团与两个相邻的碳原子结合,因此这些二醇为邻二醇。邻二醇形成缩酮中的5元环。特别合适的是选自以下的邻二醇:乙烷-1,2-二醇、丙烷-1,2-二醇、丁烷-1,2-二醇、丁烷-2,3-二醇、2-甲基丙烷-1,2-二醇、苯-1,2-二醇和同式-环己烷-1,2-二醇,特别是乙烷-1,2-二醇。其它特别合适的是这样的二醇,其中羟基基团被3个碳原子分隔,并从而形成缩酮中的非常稳定的6元环。特别合适的这种二醇为丙烷-1,3-二醇、丁烷-1,3-二醇、2-甲基丙烷-1,3-二醇、2-甲基丁烷-1,3-二醇、2,2-二甲基丙烷-1,3-二醇、1,2-二甲基丙烷-1,3-二醇、3-甲基戊烷-2,4-二醇和2-(羟甲基)环己醇。优选地,将伯醇用作二醇。用于缩酮形成的反应条件和化学计量对于本领域的技术人员而言是已知的。优选的缩酮具有式(XI)或(XII)其中波形线表示碳-碳键,其连接至相邻的碳-碳双键以使所述碳-碳双键呈Z-构型或E-构型;并且其中式中具有虚线的双键表示单碳-碳键或双碳-碳键;并且其中表示立体异构中心;并且其中Q1和Q2单独或一起表示C1-C10烷基基团或卤代C1-C10烷基基团;或一起形成C2-C6亚烷基基团或C6-C8亚环烷基基团。Q1和Q2特别表示直链C1-C10烷基基团或氟代直链C1-C10烷基基团,优选地直链C1-C4烷基基团或-CH2CF3基团或式的基团;其中Q3、Q4、Q5和Q6彼此独立地为氢原子或甲基或乙基基团。特别地,Q1和Q2一起表示氟代直链C1-C10烷基基团,优选地,直链C1-C4烷基基团或-CH2CF3基团或一起形成亚烷基基团CH2-C(CH3)2-CH2。因此,优选的待不对称氢化的缩酮选自由如下组成的组:(E)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(E)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(E)-2,6-二甲基-10,10-双(2,2,2-三氟乙氧基)十一碳-2,6-二烯、(E)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一碳-5-烯、(Z)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(Z)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(Z)-2,6-二甲基-10,10-双(2,2,2-三氟乙氧基)十一碳-2,6-二烯、(Z)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一碳-5-烯;(E)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷、(E)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯、(Z)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷和(Z)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯;(R,E)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷、(R,E)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯;(R,Z)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷和(R,Z)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯。将经氢化的缩酮水解成酮对于本领域的技术人员而言是已知的。特别合适的是通过酸水解并且分离所形成的酮,特别是通过萃取。不对称氢化步骤c)和f)包括在手性铱络合物的存在下通过分子氢进行不对称氢化。手性铱络合物是具有与中心铱原子配位的有机配体的化合物。手性铱络合物的手性归因于配体的手性或配体的空间排列。由络合物化学可熟知手性的概念。配体可为单齿配体或多齿配体。优选地,与铱中心原子结合的配体为螯合配体。对于本发明,已证实特别是具有携带立体异构中心的有机配体的手性铱络合物是非常合适的。优选的是,手性铱络合物与具有作为配位原子的N和P的螯合有机配体结合以及与两个烯烃或具有两个碳-碳双键的二烯结合,因此手性铱络合物优选地具有下式(III-0)其中P-Q-N表示包含立体异构中心的螯合有机配体或具有平面或轴向手性并且具有作为与络合物的铱中心的结合位点的氮和磷原子;Y1、Y2、Y3和Y4彼此独立地为氢原子、C1-12-烷基、C5-10-环烷基或芳族基团;或它们中的至少两个一起至少形成具有至少2个碳原子的二价桥连基团;前提条件是Y1、Y2、Y3和Y4并非全是氢原子;并且为阴离子,特别是选自卤化物、PF6-、SbF6-、四(3,5-双(三氟甲基)苯基)硼酸根(BArF-)、BF4-、全氟磺酸根,优选F3C-SO3-或F9C4-SO3-;ClO4-、Al(OC6F5)4-、Al(OC(CF3)3)4-、N(SO2CF3)2-N(SO2C4F9)2-和B(C6F5)4-。在配体P-Q-N的化学式中氮和磷原子优选地被2至5个原子,优选被3个原子分隔。螯合有机配体P-Q-N优选地选自式(III-N1)、(III-N2)、(III-N3)、(III-N4)、(III-N5)、(III-N6)、(III-N7)、(III-N8)和(III-N9)其中G1表示C1-C4-烷基、C5-7-环烷基、金刚烷基、苯基(任选地被一至三个C1-5-烷基、C1-4-烷氧基、C1-4-全氟烷基基团和/或一至五个卤素原子所取代))、苄基、1-萘基、2-萘基、2-呋喃基基团;G2、G3和G4彼此独立地表示氢原子或C1-C4-烷基、C5-7-环烷基、金刚烷基、苯基(任选地被一至三个C1-5-、C1-4-烷氧基、C1-4-全氟烷基基团和/或一至五个卤素原子所取代))、苄基、1-萘基、2-萘基、2-呋喃基基团;X1和X2彼此独立地为氢原子、C1-4-烷基、C5-7-环烷基、金刚烷基、苯基(任选地被一至三个C1-5-烷基、C1-4-烷氧基、C1-4-全氟烷基基团和/或一至五个卤素原子所取代))、苄基、1-萘基、2-萘基、2-呋喃基或二茂铁基;Ph表示苯基;n为1或2或3,优选地为1或2;并且R1、Z1和Z2如后文针对式(III)所定义如果Y1和Y2和/或Y3和Y4形成式Y1-=-Y2和/或式Y3-=-Y4的烯烃,则此烯烃或这些烯烃优选地选自由如下组成的组:乙烯、丙-1-烯、2-甲基丙-1-烯、2-甲基丁-2-烯、2,3-二甲基丁-2-烯、(Z)-环辛烯、环己烯、环庚烯、环戊烯和降冰片烯。如果Y1、Y2、Y3和Y4形成二烯,则其为环状的(双键在环中)或无环的(双键不在环中)。二烯的两个碳-碳双键优选地通过两个碳键连接,即二烯优选地包含亚结构C=C-C-C-C=C。优选的无环二烯的例子为己-1,5-二烯、庚-1,5-二烯、辛-1,5-二烯、辛-2,6-二烯、2,4-二烷基-2,7-辛二烯、3,6-二烷基辛-2,6-二烯、1,2-二乙烯基环己烷和1,3-丁二烯。环状二烯的例子为环辛-1,5-二烯、环己-1,4-二烯、环己-1,3-二烯、3,4,7,8-四烷基环辛-1,5-二烯、3,4,7-三烷基环辛-1,5-二烯、3,4-二-烷基环辛-1,5-二烯、3,7-二-烷基环辛-1,5-二烯、3,8-二-烷基环辛-1,5-二烯、3-烷基环辛-1,5-二烯;降冰片二烯、1-烷基降冰片二烯、2-烷基降冰片二烯、7-烷基降冰片二烯、二环戊二烯、环戊二烯和(1s,4s)-二环[2.2.2]辛-2,5-二烯。优选的二烯为环辛-1,5-二烯。一类高度优选的手性铱络合物为式(III)的手性铱络合物其中n为1或2或3,优选地为1或2;X1和X2彼此独立地为氢原子、C1-4-烷基、C5-7-环烷基、金刚烷基、苯基(任选地被一至三个C1-5-、C1-4-烷氧基、C1-4-全氟烷基基团和/或一至五个卤素原子所取代))、苄基、1-萘基、2-萘基、2-呋喃基或二茂铁基;Z1和Z2彼此独立地为氢原子、C1-5-烷基或C1-5-烷氧基基团;或Z1和Z2一起表示形成5至6元环的桥连基团;为阴离子,特别是选自卤素离子、PF6-、SbF6-、四(3,5-双(三氟甲基)苯基)硼酸根(BArF-)、BF4-、全氟磺酸根,优选F3C-SO3-或F9C4-SO3-;ClO4-、Al(OC6F5)4-、Al(OC(CF3)3)4-、N(SO2CF3)2-N(SO2C4F9)2-和B(C6F5)4-;R1表示苯基或邻甲苯基或间甲苯基或对甲苯基或者式(IVa)或(IVb)或(IVc)的基团其中R2和R3二者均表示H或C1-C4-烷基基团或卤代C1-C4-烷基基团,或表示一起形成6元脂环族或芳族环的二价基团,其任选地被卤素原子或被C1-C4-烷基基团或被C1-C4-烷氧基基团所取代R4和R5二者均表示H或C1-C4-烷基基团或卤代C1-C4-烷基基团,或一起形成6元脂环族或芳族环的二价基团,其任选地被卤素原子或被C1-C4-烷基基团或被C1-C4-烷氧基基团所取代;R6和R7和R8各自表示C1-C4-烷基基团或卤代C1-C4-烷基基团;R9和R10二者均表示H或C1-C4-烷基基团或卤代C1-C4-烷基基团,或一起形成6元脂环族或芳族环的二价基团,其任选地被卤素原子或被C1-C4-烷基基团或被C1-C4-烷氧基基团所取代;并且其中*表示式(III)的络合物的立体异构中心。式(III)的络合物是中性的,即所述络合物由式(III’)的络合物阳离子和阴离子Y组成,如前所述。本领域的技术人员知道阴离子和阳离子可以被解离。X1和/或X2优选地表示氢原子、甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、正戊基、异戊基、新戊基、环戊基、环己基、环庚基、金刚烷基、苯基、苄基、邻甲苯基、间甲苯基、对甲苯基、4-甲氧基苯基、4-三氟甲基苯基、3,5-二-叔丁基苯基、3,5-二甲氧基苯基、1-萘基、萘基、2-呋喃基、二茂铁基或被一至五个卤素原子取代的苯基基团。在X1和/或X2表示被一至五个卤素原子取代的苯基基团的情况下,被氟原子取代的苯基基团是特别有用的,即C6H4F、C6H3F2、C6H2F3、C6HF4或C6F5。在X1和/或X2表示被一至三个C1-4-烷基取代的苯基基团的情况下,被一个或多个甲基基团取代的苯基基团是特别有用的,特别是邻甲苯基和对甲苯基。优选地,X1和X2表示相同的取代基。最优选地,X1和X2均为苯基或邻甲苯基基团。优选的是,在上述R2、R3、R4、R5、R6、R7、R8、R9和R10的定义中使用的C1-C4-烷基或烷氧基基团为伯或仲(优选为伯)烷基或烷氧基基团。式(IVa)的特别合适的取代基R1为9-蒽基或1-萘基基团。式(IVb)的另一个特别合适的取代基R1为2,4,6-三甲苯基(mesityl)基团。式(IVc)的另一个特别合适的取代基R1为2-萘基基团。优选地,R1由苯基(缩写为“Ph”)或式(IV-1)或(IV-2)或(IV-3),特别是(IV-1)或(IV-3)表示。已发现,最优选的取代基R1为9-蒽基或苯基。优选的式(III)的手性铱络合物为式(III-A)、(III-B)、(III-C)、(III-D)、(III-E)和(III-F)的络合物。最优选的作为式(III)的手性铱络合物的是式(III-C)和(III-D)和(III-F)的络合物,特别是式(III-C)或(III-F)的络合物。式(III)的手性铱络合物可如Chem.Sci.,2010,1,72-78中详述来相应地合成,该文献的全部内容通过引用结合于此。式(III)的铱络合物为手性的。由星号标记的所述手性中心的手性为S或R,即存在式(III)的手性络合物的两种对映体(IIIa)和(IIIb):式(III)的络合物的各个对映体可以在络合步骤之后大部分从外消旋混合物分离。然而,如Chem.Sci.,2010,1,72-78所公开,式(III)的络合物的合成包括涉及非外消旋手性醇的反应。由于已知另外的反应步骤不改变所述络合物的手性,因此其异构体纯度(S:R比率)取决于所述醇的对映体纯度。由于所述相应的醇可分别以大于99%和低于1%的R/S比率获得,式(III)的络合物可以极高的对映体纯度获得,特别是分别以大于99%和低于1%的R/S比率获得。优选地,以一种对映体过量地使用手性铱络合物。具体而言,优选的是式(III)的手性铱络合物的各个对映体的摩尔量的比率R:S大于90:10或小于10:90,优选地在100:0至98:2或0:100至2:98的范围内。最优选的是,所述比率分别为约100:0和约0:100。最终优选比率分别为100:0和0:100。在一个实施方案中,由*指示的立体异构中心具有R-构型。在另一个实施方案中,由*指示的立体异构中心具有S-构型。氢化剂为分子氢(H2)。分别基于酮和缩酮的量,手性铱络合物的量优选为约0.0001至约5摩尔%,优选为约0.001至约2摩尔%,更优选为约0.01至约1摩尔%。氢化可以本体进行或在惰性载体中进行,特别是在惰性溶剂或惰性溶剂的混合物中进行。优选地,氢化以本体(纯的)进行。优选的合适溶剂为卤代烃、烃、碳酸酯、醚和卤代醇。特别优选的溶剂为烃、氟化醇和卤代烃,特别是卤代脂族烃。烃的优选例子为己烷、庚烷、甲苯、二甲苯和苯,特别是甲苯和庚烷。优选的醚为二烷基醚。特别有用的醚为具有小于8个碳原子的二烷基醚。最优选的醚为甲基叔-丁基醚(CH3-O-C(CH3)3)。优选的卤代醇为氟化醇。特别优选的氟化醇为2,2,2-三氟乙醇。一类优选的卤代烃为卤代芳族化合物,特别是氯苯。卤代脂族烃的优选例子为单或多卤代的直链或支链或环状的C1-至C15-烷烃。尤其优选的例子为单或多氯化或溴化的直链或支链或环状的C1-至C15-烷烃。更优选的是单或多氯化的直链或支链或环状的C1-至C15-烷烃。最优选的是二氯甲烷、1,2-二氯乙烷、1,1,1-三氯乙烷、氯仿和二溴甲烷。用于氢化的最优选试剂为二氯甲烷。所用溶剂的量并非很关键。然而,已证实,待氢化的酮或缩酮的浓度优选地介于0.05和1M之间,特别是介于0.2和0.7M之间。氢化反应适宜地在约1至约100巴的分子氢绝对压力下,优选在约20至约75巴的分子氢绝对压力下进行。反应温度适宜地介于约0至约100℃之间,优选地介于约10至约60℃之间。反应物和溶剂的添加顺序并非关键。适用于氢化的技术和装置对于本领域的技术人员而言大体上是已知的。通过不对称氢化,前手性碳-碳双键被氢化以在一个或两个碳原子处形成手性立体异构中心。在步骤c)中,6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮,或6,10-二甲基十一碳-5-烯-2-酮的缩酮或6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮被不对称氢化。在步骤f中,6,10,14-三甲基十五碳-5-烯-2-酮或6,10,14-三甲基十五碳-5-烯-2-酮的缩酮被氢化。如果缩酮被不对称氢化,则在不对称氢化后,经不对称氢化的缩酮优选地具有式(XV)或(XVI)。其中表示立体异构中心;并且其中Q1和Q2如针对式(XI)和(XII)所定义。因此,已被不对称氢化的优选的缩酮优选地选自由如下组成的组:2-(4,8-二甲基壬基)-2,5,5-三甲基-1,3-二噁烷、6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一烷、2,5,5-三甲基-2-(4,8,12-三甲基十三基)-1,3-二噁烷、6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷;(R)-2-(4,8-二甲基壬基)-2,5,5-三甲基-1,3-二噁烷、(R)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一烷、2,5,5-三甲基-2-((4R,8R)-4,8,12-三甲基十三基)-1,3-二噁烷和(6R,10R)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷。当这些缩酮被水解成相应的酮时,它们分别生成6,10-二甲基十一烷-2-酮或6,10,14-三甲基十五烷-2-酮,或(R)-6,10-二甲基十一烷-2-酮或(6R,10R)-6,10,14-三甲基十五烷-2-酮。尽管在手性铱络合物(特别是式(III)的那些)的存在下通过分子氢对6,10-二甲基十一碳-5-烯-2-酮、6,10-二甲基十一碳-5,9-二烯-2-酮和(R)-6,10,14-三甲基十五碳-5-烯-2-酮进行的不对称氢化已相当快速和有效并且表现出高转化率以及优异的选择性,但已观察到,当相应的酮的缩酮被不对称氢化时,不对称氢化甚至还可以得到改善。已观察到,具有特定手性(R或S)的手性铱络合物将起始物料转化成带有特定立体异构中心的产物,所述产物由于不对称氢化而形成。由于在本发明中希望通过不对称氢化产生带有具备R-构型的立构中心的产物,即分别为在步骤c)中的(R)-6,10-二甲基十一烷-2-酮和在步骤f)中的(6R,10R)-6,10,14-三甲基十五烷-2-酮,因此需要根据分别在步骤c)和步骤e)中被分离的烯烃异构体是否具有Z-或E-构型来对手性铱络合物的手性进行选择。已证实,当在由*指示的立体异构中心处具有S-构型的式(III)的手性铱络合物用于氢化E-异构体,即分别为(E)-6,10-二甲基十一碳-5-烯-2-酮和(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮时,获得在新形成的立体异构中心处带有R-构型的相应产物,即在步骤c)中的(R)-6,10-二甲基十一烷-2-酮和在步骤f)中的(6R,10R)-6,10,14-三甲基十五烷-2-酮。相应地,在由*指示的立体异构中心处具有R-构型的式(III)的手性铱络合物的存在下,对Z-异构体(即分别为(Z)-6,10-二甲基十一碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮)进行氢化提供相同的产物,即获得在新形成的立体异构中心处带有R-构型的在步骤c)中的(R)-6,10-二甲基十一烷-2-酮和在步骤f)中的(6R,10R)-6,10,14-三甲基十五烷-2-酮。意外的是,已发现该结果与是否在步骤c)或f)中使用酮或缩酮无关。因此,用于不对称氢化的步骤c)和/或f)中的式(III)的手性铱络合物优选地具有如下特征:当(E)-6,10-二甲基十一碳-5-烯-2-酮、(E)-6,10-二甲基十一碳-5,9-二烯-2-酮或其缩酮,或(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮或其缩酮待被氢化时,式(III)的手性铱络合物在由*指示的立体异构中心处具有S-构型;或当(Z)-6,10-二甲基十一碳-5-烯-2-酮、(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮或其缩酮,或(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮或其缩酮待被氢化时,式(III)的手性铱络合物在由*指示的立体异构中心处具有R-构型;添加剂在本发明的一个优选实施方式中,步骤c)和/或步骤f)中的不对称氢化在添加剂的存在下进行,所述添加剂选自有机磺酸、有机磺酸的过渡金属盐、金属醇盐、铝氧烷、烷基铝氧烷和B(R)(3-v)(OZ)v;其中v表示0、1、2或3,R表示F、C1-6-烷基、卤代C1-6-烷基、芳基或卤代芳基基团;并且Z表示C1-6-烷基、卤代C1-6-烷基、芳基或卤代芳基基团。特别合适的添加剂选自由如下组成的组:三氟甲磺酸、烷基铝氧烷,特别是甲基铝氧烷、乙基铝氧烷、四烷氧基钛酸酯、B(R)(3-v)(OZ)v;特别是三-异丙基硼酸酯和三乙基硼烷以及BF3(优选地以BF3醚合物的形式)。作为有机磺酸的过渡金属盐特别有用的是有机磺酸的钪、铟、钇和锆盐。金属醇盐对于本领域的技术人员而言是已知的。此术语特别是指周期系的4族和13族的元素的醇盐。对于本领域的技术人员而言也已知的是,金属醇盐通常不形成良好界定的结构。在特性上,金属醇盐具有通过氧原子与金属中心结合的烃基基团。金属醇盐也可以具有由氧或含氧基团桥连的不同金属中心,例如(多核)铝氧代醇盐。作为金属醇盐特别有用的是钛醇盐(也称为烷氧基钛酸酯/盐)、锆醇盐(也称为烷氧基锆酸酯/盐)或铝醇盐。一类特别优选的金属醇盐具有多核铝氧代醇盐的类型,例如J.Chem.Soc.,DaltonTrans.,2002,259-266或Organometallics1993,12,2429-2431中所公开。烷基铝氧烷为特别适合用作齐格勒-纳塔类型的烯烃聚合的助催化剂的已知产物。它们通过三烷基铝化合物,特别是三甲基铝或三乙基铝的受控水解来制备。所述水解可例如通过水合金属盐(含有结晶水的金属盐)来实现。优选地,添加剂选自由如下组成的组:三氟甲磺酸、烷基铝氧烷,特别是甲基铝氧烷、乙基铝氧烷、四烷氧基钛酸酯、B(R)(3-v)(OZ)v;特别是三-异丙基硼酸酯和三乙基硼烷以及BF3(优选地以BF3醚合物形式)。更优选的是三氟甲磺酸、烷基铝氧烷,特别是甲基铝氧烷、乙基铝氧烷、四烷氧基钛酸酯、B(R)(3-v)(OZ)v;特别是三-异丙基硼酸酯和三乙基硼烷。尤其良好的结果已通过如下添加剂获得,所述添加剂已从三甲基铝氧烷和2,2,2-三氟乙醇中或从三烷基铝和2,2,2-三氟乙醇中获得。已发现,当使用上述添加剂时,在手性铱络合物的存在下使用分子氢进行的不对称氢化的质量和速度显著提高。已进一步观察到,最显著的是,当上述添加剂与待不对称氢化的酮的相应缩酮(即6,10-二甲基十一碳-5-烯-2-酮、6,10-二甲基十一碳-5,9-二烯-2-酮和(R)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮)一起使用时,不对称氢化的效率最大化。提高的效率具有以下效果,手性铱络合物的量可通过使用6,10-二甲基十一碳-5-烯-2-酮、6,10-二甲基十一碳-5,9-二烯-2-酮或(R)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮和/或通过添加上述添加剂(特别是与氟化醇、特别是2,2,2-三氟乙醇组合)而被显著降低,从而在与如同所述的6,10-二甲基十一碳-5-烯-2-酮、6,10-二甲基十一碳-5,9-二烯-2-酮和(R)-6,10,14-三甲基十五碳-5-烯-2-酮的相应不对称氢化相比的不对称氢化中实现给定的收率和立体特异性氢化。当上述方法包括如上详述的顺式/反式异构化的步骤时,本发明的方法极具吸引力,因为对于所有起始物料的最佳使用,不必针对使用相反手性的氢化络合物对每个异构体进行的单独不对称氢化设置两条平行生产线。因此,非常优选的是如上所述的原位异构化。化学转化在步骤d)中,将(R)-6,10-二甲基十一烷-2-酮化学转化成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物;该化学转化可以不同的方式进行。在一个优选的方法中,在步骤d)中,将(R)-6,10-二甲基十一烷-2-酮至(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物的化学转化通过以下步骤进行:要么d1)在碱性物质的存在下使用乙炔对(R)-6,10-二甲基十一烷-2-酮进行乙炔化以生成(7R)-3,7,11-三甲基十二碳-1-炔-3-醇;d2)在林德拉催化剂的存在下用分子氢对(7R)-3,7,11-三甲基十二碳-1-炔-3-醇进行氢化以生成(7R)-3,7,11-三甲基十二碳-1-烯-3-醇;要么d1’)通过添加乙烯基格氏试剂(Grignardreagent)对(R)-6,10-二甲基十一烷-2-酮进行乙烯化以生成(7R)-3,7,11-三甲基十二碳-1-烯-3-醇;然后是要么d3)将(7R)-3,7,11-三甲基十二碳-1-烯-3-醇与2-甲氧基丙-1-烯反应以生成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物;要么d3’)在碱和/或酸的存在下将(7R)-3,7,11-三甲基十二碳-1-烯-3-醇与乙酰乙酸烷基酯或双烯酮反应以生成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物。使用步骤d1)的变型的反应类型和条件的细节公开于EP1532092B1中(特别是实施例2中),或WO2003/029175A1(使用碱性阴离子交换树脂)中,所述专利的全部内容通过引用结合于此。在步骤d2)中在林德拉催化剂(Lindlarcatalyst)的存在下用分子氢进行氢化对于本领域的技术人员而言是已知的。例如,A.Ofner等人,Helv.Chim.Acta1959,42,2577-2584公开了步骤d1)和d2)的组合,该文献的全部内容通过引用结合于此。例如,US4,028,385公开了使用步骤d1’)的变型的反应类型和条件的细节以及针对步骤d1)和d2)的细节,该专利的全部内容通过引用结合于此。使用步骤d3)(Saucy-Marbet反应)的变型的反应类型和条件的细节公开于例如DE1193490和DE19649564A1中,这两篇文献的全部内容通过引用结合于此。步骤d3’)(卡莱尔重排)中第一种变型的反应类型和条件的细节公开于例如TheClaisenRearrangement的第8章“TheCarrollRearrangement”(Hatcher等人)中;Hiersemann,M.;Nubbemeyer,U.,Eds.;Wiley-VCHVerlagGmbH&Co.KGaA,2007;397-430,这两篇文献的全部内容通过引用结合于此。在卡莱尔重排中,在碱(优选乙酸碱金属盐,例如乙酸钠)的存在下,用乙酰乙酸烷基酯(优选乙酰乙酸甲酯或乙酰乙酸乙酯)进行反应。步骤d3’)中第二种变型的反应类型和条件公开于例如US2,795,617或W.Kimel等人,J.Org.Chem.1957,22,1611-1618中,这两篇文献的全部内容通过引用结合于此。分别在优选地吡啶和乙酸的存在下以及在醇盐的存在下,优选地用双烯酮进行上述反应。如早先所述,(6R,10R)-6,10,14-三甲基十五烷-2-酮是一种重要的中间体并且特别可用于合成(R,R)-异植醇、(2-双型)-α-生育酚或(2R,4’R,8’R)-α-生育酚。因此,在另一个方面,本发明涉及一种制备(R,R)-异植醇((3RS,7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇)的方法,其包括:如上详述的制备(6R,10R)-6,10,14-三甲基十五烷-2-酮的方法;随后的步骤:要么g)在碱的存在下使用乙炔对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙炔化以生成(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇;h)在林德拉催化剂的存在下用分子氢对(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇进行氢化以生成(R,R)-异植醇;要么h’)通过添加乙烯基格氏试剂对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙烯化以生成(R,R)-异植醇。步骤g)和h)和h’)的条件对应于针对步骤d)中类似的步骤d1)和d2)和d1’)所描述的条件。在另一个方面,本发明涉及一种制备式(V)的化合物的方法,其包括:如上详述的制备(6R,10R)-6,10,14-三甲基十五烷-2-酮的方法;随后的步骤:要么g)在碱性物质的存在下使用乙炔对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙炔化以生成(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇;h)在林德拉催化剂的存在下用分子氢对(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇进行氢化以生成(R,R)-异植醇;要么h’)通过添加乙烯基格氏试剂对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙烯化以生成(R,R)-异植醇;随后的步骤:m)将(R,R)-异植醇与式(VI)的化合物缩合以生成式(V)的化合物,所述式(V)的化合物对由#指示的中心处的手性而言是异构混合物;其中#表示立体异构中心。步骤g)和h)和h’)的条件对应于针对步骤d)中类似的步骤d1)和d2)和d1’)所描述的条件。如步骤m)所描述的(R,R)-异植醇与式(VI)的化合物的缩合反应对于本领域的技术人员而言是已知的。对于这种缩合,可以使用一系列催化剂,例如ZnCl2/无机酸、BF3/AlCl3、Fe/HCl、三氟乙酸或硼酸/羧酸以及铟(III)或钪(III)盐,如WO2005/121115A1中所公开。此外,合适的催化剂为杂多酸,特别是12-钨磷酸或12-钨硅酸,例如EP0970953A1中所公开。式(V)的化合物表示(2-双型)-α-生育酚,即对应的(2R,4’R,8’R)-α-生育酚和(2S,4’R,8’R)-α-生育酚)的混合物。在另一个方面,本发明涉及一种制备式(V-A)的化合物的方法,其包括:如上详述的制备(6R,10R)-6,10,14-三甲基十五烷-2-酮的方法;随后的步骤:要么g)在碱性物质的存在下使用乙炔对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙炔化以生成(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇;h)在林德拉催化剂的存在下用分子氢对(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇进行氢化以生成(R,R)-异植醇;要么h’)通过添加乙烯基格氏试剂对(6R,10R)-6,10,14-三甲基十五烷-2-酮进行乙烯化以生成(R,R)-异植醇;随后的步骤:m)将(R,R)-异植醇与式(VI)的化合物缩合以生成式(V)的化合物,所述式(V)的化合物对由#指示的中心处的手性而言是异构混合物;其中#表示立体异构中心;和n)从式(V)的异构混合物分离式(V-A)的化合物除了附加步骤n)之外,这种制备式(V-A)的化合物的方法与制备式(V)的化合物的方法相同。将(2R,4’R,8’R)-α-生育酚从对应的(2-双型)-α-生育酚分离可借助于手性相通过色谱分离来实现,特别是如WO2012/152779A1中所述。也优选的是,通过(2S,4’R,8’R)-α-生育酚中富含的级份的差向异构(epimerization)来提高(2R,4’R,8’R)-α-生育酚的收率,如WO2012/152779A1中步骤c)所公开。WO2012/152779A1的全部内容通过引用结合于此。选自如下组的物质对于生育酚、维生素K1的合成以及对于调味剂和芳香剂或药品而言是重要的中间体,所述组为:(E)-6,10-二甲基十一碳-5-烯-2-酮、(Z)-6,10-二甲基十一碳-5-烯-2-酮、(E)-6,10-二甲基十一碳-5,9-二烯-2-酮、(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮、(R)-6,10-二甲基十一烷-2-酮;(E)-6,10-二甲基十一碳-5-烯-2-酮的缩酮、(Z)-6,10-二甲基十一碳-5-烯-2-酮的缩酮、(E)-6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮、(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮;(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮、(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮、(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮、(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮、(6R,10R)-6,10,14-三甲基十五烷-2-酮、(6R,10R)-6,10,14-三甲基十五烷-2-酮的缩酮、(7R)-3,7,11-三甲基十二碳-1-炔-3-醇、(7R)-3,7,11-三甲基十二碳-1-烯-3-醇和(R,R)-异植醇。它们中的大部分具有典型的气味,这使其极具吸引力而被用作调味剂和芳香剂行业中产品的成分,例如香料中的成分。在另一个方面,本发明涉及一种组合物,其包含:-至少一种式(XI)或(XII)的缩酮,和-至少一种手性铱络合物。式(XI)或(XII)的缩酮和手性铱络合物、其比率以及其优选的实施方案、性质和效果已在本文件中详述。在最后一个方面,本发明涉及式(XX-A)或(XX-B)或(XX-C)或(XX-D)的缩酮其中上式中具有虚线的双键表示单碳-碳键或双碳-碳键;并且其中波形线表示碳-碳键,其连接至相邻的单碳键(表示)或连接至相邻的碳-碳双键(表示)以使所述碳-碳双键呈Z-构型或E-构型。最优选的是式(XX-B)和(XX-D)的缩酮。优选的缩酮选自由如下组成的组:(E)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(E)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(E)-2,6-二甲基-10,10-双(2,2,2-三氟乙氧基)十一碳-2,6-二烯、(E)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一碳-5-烯、(Z)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(Z)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷、(Z)-2,6-二甲基-10,10-双(2,2,2-三氟乙氧基)十一碳-2,6-二烯、(Z)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一碳-5-烯;(E)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷、(E)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯、(Z)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷和(Z)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯;(R,E)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷、(R,E)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯;(R,Z)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷和(R,Z)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯;或选自如下组成的组:2-(4,8-二甲基壬基)-2,5-二甲基-1,3-二噁烷、6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一烷、2,5,5-三甲基-2-(4,8,12-三甲基十三基)-1,3-二噁烷、6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷;(R)-2-(4,8-二甲基壬基)-2,5-二甲基-1,3-二噁烷、(R)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一烷、2,5,5-三甲基-2-((4R,8R)-4,8,12-三甲基十三基)-1,3-二噁烷和(6R,10R)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷。所有这些缩酮特别适用于如上详述的不对称氢化或为所述不对称氢化的产物。也如前所述,与对应的酮相比,不饱和酮的缩酮表现得极其有利。附图在下面的段落中,通过示意图1至6对本发明的一些优选实施方式进行进一步论述。然而,这不应理解为将本发明限于本文附图中所述的实施方式。图1至3示出从6,10-二甲基十一碳-5-烯-2-酮和6,10-二甲基十一碳-5,9-二烯-2-酮二者至(6R,10R)-6,10,14-三甲基十五烷-2-酮的后续步骤。图1至4示出从6,10-二甲基十一碳-5-烯-2-酮和6,10-二甲基十一碳-5,9-二烯-2-酮二者分别至(R,R)-异植醇、(2-双型)-α-生育酚和(2R,4’R,8’R)-α-生育酚的后续步骤。附图括号中的参考符号例如(R-II)是出于如下文所述的标识目的,并且不应与文件中其余部分中所用的式的标识例如(II)混淆。在图1中,示意性地示出合成(R)-6,10-二甲基十一烷-2-酮(R-II)的三种不同的可能性(图1a)、1b)、1c))。作为图1中示出的所有可能性的第一步骤a),提供(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的混合物或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮和(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮的混合物(E/Z-I)。在图1a)中,E-异构体(E-I)(即,分别为(E)-6,10-二甲基十一碳-5-烯-2-酮或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮)和对应的Z-异构体(Z-I)(即,分别为(Z)-6,10-二甲基十一碳-5-烯-2-酮或(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮)在步骤b)中从步骤a)中提供的混合物分离。步骤b)中的分离优选地经由柱通过蒸馏来进行。在步骤c)中,用特定的手性铱络合物使Z-异构体不对称氢化,而用相应的对映体手性铱络合物使E-异构体不对称氢化。优选的手性铱络合物为式(III)中的一者。在式(IIIa)的手性铱络合物(S-Ir-络合物)的存在下使用分子氢使E-异构体(E-I)不对称氢化,所述手性铱络合物在式(III)中*所指示的立体异构中心处具有S-构型。另一方面,在式(IIIb)的手性铱络合物(R-Ir-络合物)的存在下使用分子氢使Z-异构体(Z-I)不对称氢化,所述手性铱络合物在式(III)中*所指示的立体异构中心处具有R-构型。两种不对称氢化路线提供相同的产物,即(R)-6,10-二甲基十一烷-2-酮(R-II)。6,10-二甲基十一碳-5,9-二烯-2-酮的9位处的双键也在不对称氢化期间被氢化。然而,由于此双键不是前手性的,因此在氢化期间在此位置无手性中心形成。在图1b)中,异构体中的仅一种(E-异构体(E-I))(期望的异构体)被不对称氢化,如以上针对图1a)所描述。在步骤α)中通过添加顺式/反式异构化催化剂(c/t-cat)和加热使另一种异构体(Z-异构体(Z-I))(非期望的异构体)经受顺式/反式异构化。优选使用的顺式/反式异构化催化剂为聚硫醇,特别是式(X)的聚硫醇。通过顺式/反式异构化催化剂的作用,将(Z-异构体(Z-I))被异构化成E-异构体和Z-异构体的混合物(E/Z-I),所述混合物可在步骤β)中添加至步骤α)中提供的混合物中。图1b)示出其中在E-异构体是期望的异构体(即被不对称氢化的异构体)的情况下的过程。显然,在Z-异构体是期望的异构体(即被不对称氢化的异构体)的情况下,异构化过程将以类似的方式适于E-异构体的异构化过程。在图1c)中,异构体中的仅一种(Z-异构体(Z-I))(期望的异构体)被不对称氢化,如以上针对图1a)所描述。将顺式/反式异构化催化剂(c/t-cat)添加至步骤a)中提供的E-异构体和Z-异构体(E/Z-I)的混合物中。在步骤b)中,(期望的)异构体(Z-异构体(Z-I))的分离在顺式/反式异构化催化剂的存在下(一锅异构化或原位异构化)通过蒸馏来进行。由于期望的异构体是通过蒸馏分离,因此富含高沸点异构体的残余物被异构化,使得在蒸馏容器中连续地形成Z-异构体和E-异构体之间的热力学平衡。此程序可允许步骤a)中提供的初始存在于异构体混合物中的全部非期望的异构体均转化成期望的异构体。如所述,图1c)示出待成为期望的异构体(即被分离和不对称氢化)的Z-异构体,然而,显然以上论述也类似地适用于E-异构体将成为期望的异构体的情况。图2示出后续步骤d)。在步骤d)中,(R)-6,10-二甲基十一烷-2-酮(R-II)被化学转化成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(E/Z-R-III)的混合物。图2还示出此类化学转化的优选变型。在图2示出的变型之一中,(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(R-IIb)由(R)-6,10-二甲基十一烷-2-酮(R-II)形成,方式是通过在第一步骤即步骤d1)中使(R)-6,10-二甲基十一烷-2-酮(R-II)与乙炔(电石气)在碱(示出的是KOH)的存在下进行反应以生成中间体(7R)-3,7,11-三甲基十二碳-1-炔-3-醇(R-IIa),然后在第二步骤即步骤d2)中与分子氢在林德拉催化剂的存在下进行反应。在另一变型中,(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(R-IIb)通过与格氏试剂(Grignardreagent)反应而被直接形成。在图2中,示出作为格氏试剂的乙烯基氯化镁。随后,用于将(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(R-IIb)转化成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(E/Z-R-III)的混合物的两种变型在图2中示出。在第一种变型中,(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(R-IIb)与2-甲氧基丙-1-烯进行Saucy-Marbet反应(步骤d3))以生成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(E/Z-R-III)的混合物。在第二种变型中,(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(R-IIb)与乙酰乙酸烷基酯(优选乙酰乙酸甲基酯)在碱(优选醋酸钠)的存在下进行反应以生成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(E/Z-R-III)的混合物(卡莱尔重排)。图3示出后续步骤e)和f)。图3对应于图1,不同的是个别物质被C5单元扩展。类似地,(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物(E/Z-R-III)的至少一种异构体在步骤e)中被分离并且在步骤f)中被不对称氢化成(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV)。图4示出由(6R,10R)-6,10,14-三甲基十五烷-2-酮分别至(R,R)-异植醇、(2-双型)-α-生育酚和(2R,4’R,8’R)-α-生育酚的后续步骤。图4示出用于将(6R,10R)-6,10,14-三甲基十五烷-2-酮转化成(R,R)-异植醇的两种变型。在第一种变型中,(R,R)-异植醇(R-V)由(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV)形成,方式是通过在第一步骤即步骤g)中使(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV)与乙炔(电石气)在碱(示出的是KOH)的存在下进行反应以生成中间体(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇(R-IVa),然后在第二步骤即步骤h)中与分子氢在林德拉催化剂的存在下进行反应。在示出的另一变型中,(R,R)-异植醇(R-V)通过与格氏试剂反应由(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV)形成。在图4中,示出作为格氏试剂的乙烯基氯化镁。(R,R)-异植醇(R-V)可进一步在步骤m)中与2,3,5-三甲基苯-1,4-二醇缩合生成(2-双型)-α-生育酚(R/S-VI))。在下一步骤n)中,将(2R,4’R,8’R)-α-生育酚(R-VI))从相应的(2-双型)-α-生育酚(R/S-VI)分离。分离优选地借助于手性相通过色谱分离来进行。在图5和6中,示出不对称氢化的优选实施方案。图5涉及图1中的工艺步骤,图6涉及图3中的工艺步骤。图5的左侧示出在步骤c0)中使用醇(图5中示出乙二醇)在酸的存在下分别形成(E)-6,10-二甲基十一碳-5-烯-2-酮或(E)-6,10-二甲基十一碳-5,9-二烯-2-酮(E-I)的缩酮(E-IK)(其在步骤b)中分离对应的异构体混合物后得到)。缩酮(E-IK),优选(E)-2-(4,8-二甲基壬-3-烯-1-基)-2-甲基-1,3-二氧戊环或(E)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2-甲基-1,3-二氧戊环,随后分别在步骤c)中被不对称氢化,如图1中所讨论。此不对称氢化的直接产物为经不对称氢化的缩酮,即优选的(R)-2-(4,8-二甲基壬基)-2-甲基-1,3-二氧戊环(R-IIK),其在步骤c’)中酸性水解后生成(R)-6,10-二甲基十一烷-2-酮(R-II)。在图5的右侧,示出用于Z-异构体的相应反应方案,即(Z)-6,10-二甲基十一碳-5-烯-2-酮或(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮(Z-I),分别经由相应的缩酮中间体提供相同化合物(R)-6,10-二甲基十一烷-2-酮(R-II),优选地缩酮中间体分别为(Z)-2-(4,8-二甲基壬-3-烯-1-基)-2-甲基-1,3-二氧戊环或(Z)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2-甲基-1,3-二氧戊环(Z-IK)。图6的左侧示出在步骤fo)中形成(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮(E-R-III)的缩酮(E-R-IIIK),其在步骤e)中的异构体分离后使用醇(图6中示出乙二醇)在酸的存在下获得。缩酮(E-R-IIIK),优选(R,E)-2-甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二氧戊环,随后在步骤f)中被不对称氢化,如图3中所讨论。此不对称氢化的直接产物为经不对称氢化的缩酮,即2-甲基-2-((4R,8R)-4,8,12-三甲基十三基)-1,3-二氧戊环(R-IVK),其在步骤f’)中酸性水解后生成(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV)。在图6的右侧,示出用于Z-异构体的相应反应方案,即(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(Z-R-III),经由缩酮中间体提供相同化合物(6R,10R)-6,10,14-三甲基十五烷-2-酮(R-IV),优选地缩酮中间体为(Z,E)-2-甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二氧戊环(Z-R-IIIK)。实施例通过以下实验对本发明进行进一步说明。分析方法6,10-二甲基十一碳-5-烯-2-酮(DHGA)、(R)-6,10-二甲基十一烷-2-酮(THGA)和(R)-6,10,14-三甲基十五碳-5-烯-2-酮(R-THFA)的E/Z-比率和/或纯度的GC测定:Agilent6850,柱DB-5HT(30m,0.25mm直径,0.10μm膜厚),107kPa氦载气)。样品以己烷中的溶液的形式被进样,分流比300:1,进样器温度200℃,检测器温度350℃。烘箱温度程序:100℃(8min),10℃/min至200℃(1min),20℃/min至220℃(4min),运行时间24min。(6R,10R)-6,10,14-三甲基十五烷-2-酮纯度的GC测定Agilent6850,柱DB-5HT(30m,0.25mm直径,0.10μm膜厚),115kPa氦载气)。样品以己烷中的溶液的形式被进样,分流比300:1,进样器温度200℃,检测器温度350℃。烘箱温度程序:120℃(5min),14℃/min至260℃(2min),20℃/min至280℃(4min),运行时间22min。(3RS,7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇((R,R)-异植醇)纯度的GC测定配备有FID的Agilent6850仪器。AgilentDB-5柱(30m,0.32mm直径,0.25μm膜厚),具有25psi分子氢载气。样品以乙腈中的溶液的形式被进样,分流比为50:1。进样器温度:250℃,检测器温度:350℃。烘箱温度程序:100℃,4℃/min至250℃。6,10-二甲基十一碳-5,9-二烯-2-酮和缩酮的E/Z-比率和/或纯度的GC测定:Agilent6850仪器,柱AgilentDB-5(123-5032E,30m×0.32mm,膜0.25μm),样品以乙腈中的溶液的形式被进样,分流比为50:1,进样器250℃,检测器350℃。烘箱温度程序:100℃,4℃/min直至250℃,37.5min总运行时间。保留时间(tR):min.(E)-6,10-二甲基十一碳-5,9-二烯-2-酮(E-GA)11.0E-GA-DM14.8E-GA-neo20.5E-GA-tfe13.2,pc1(Z)-6,10-二甲基十一碳-5,9-二烯-2-酮(Z-GA)10.6Z-GA-DM14.0,pc1Z-GA-neo19.5E-DHGA-DM14.1,pc1E-DHGA-neo19.6,pc1E-DHGA-tfe12.5Z-DHGA-DM13.0,pc1Z-DHGA-neo18.5,pc1R,E-THFA-DM24.2,pc1R,E-THFA-neo29.1R,Z-THFA-DM23.1,pc1R,Z-THFA-neo27.9R-THGA-DM13.1R-THGA-neo18.9R-THGA-tfe11.8RR-C18-DMdecomp.2RR-C18-neo28.5RR-C18-tfe21.41pc=部分降解2decomp.=GC分析期间降解经不对称氢化的反应产物的分析相应的二甲基、乙二醇、新戊基和双(三氟乙基)缩酮在含水酸的存在下水解成酮,然后使用以下针对酮的方法分析转化率及其立体异构体比率。氢化反应的转化率使用非手性柱通过气相色谱法来测定。用于转化的方法:配备有FID的Agilent7890AGC。AgilentHP-5柱(30m,0.32mm直径,0.25μm膜厚),具有25psi分子氢载气。样品以二氯甲烷中的溶液的形式被进样,分流比为10:1。进样器温度:250℃,检测器温度:300℃。烘箱温度程序:50℃(2min),然后15℃/min至300℃,保持5min。对于异构体比率的测定,可使经氢化的酮与(+)-二异丙基-O,O’-双(三甲基甲硅烷基)-L-酒石酸酯或(-)-二异丙基-O,O’-双(三甲基甲硅烷基)-D-酒石酸酯在三氟甲磺酸三甲基甲硅烷基酯[Si(CH3)3(OSO2CF3)]的存在下反应以形成非对映体缩酮,如A.Knierzinger,W.Walther,B.Weber,R.K.Müller,T.Netscher,Helv.Chim.Acta1990,73,1087-1107中所述。使用非手性柱通过气相色谱法来分析缩酮,以测定异构体比率。对于经氢化的酮6,10-二甲基十一烷-2-酮,可使用D-(-)-或L-(+)-二异丙基酒石酸酯。对于6,10,14-三甲基十五烷-2-酮,可用L-(+)-二异丙基酒石酸酯来测量存在的(6R,10R)-异构体的量。可用D-(-)-二异丙基酒石酸酯来测定(6S,10S)-异构体的量。因此,可间接测定立体选择性氢化的选择性。测定异构体的方法:配备有FID的Agilent6890NGC。AgilentCP-Sil88,FAME柱(60m,0.25mm直径,0.20μm膜厚),具有16psi分子氢载气。样品以乙酸乙酯中的溶液的形式被进样,分流比为5:1。进样器温度:250℃,FID检测器温度:250℃。烘箱温度程序:165℃(等温,240min)下列实验中指示的Ir络合物根据Chem.Sci.,2010,1,72–78中的公开内容来制备。实验E1:6,10-二甲基十一碳-5-烯-2-酮的E/Z异构体混合物的分离(步骤b)根据DE1193490的实施例10制备7.02kg6,10-二甲基十一碳-5-烯-2-酮,并且根据上文给出的方法进行分析,其为(E)-6,10-二甲基十一碳-5-烯-2-酮和(Z)-6,10-二甲基十一碳-5-烯-2-酮的57%/43%混合物(99%纯度)。使用由蒸馏器(容积:9升)、降膜蒸发器、精馏柱(70mm内径,高度5m)组成的分离设备来对混合物进行蒸馏。柱配备有非常高效的规整填料(Sulzer)。在大约5毫巴的顶部压力、105℃至112℃范围内的柱顶部温度以及约125℃的蒸馏器底部温度下对混合物进行精馏。回流比率调节至20。收集包含(Z)-6,10-二甲基十一碳-5-烯-2-酮(Z-异构体的含量=99%,E-异构体<1%)的馏分以及包含(E)-6,10-二甲基十一碳-5-烯-2-酮(E-异构体的含量=97%,Z-异构体<3%)的馏分。最后,发现(E)-6,10-二甲基十一碳-5-烯-2-酮(E-异构体的含量=99.5%,Z-异构体=0.5%)留在蒸馏器中。实验E2:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的不对称氢化(步骤c)两种异构体(E)-6,10-二甲基十一碳-5-烯-2-酮(E-DHGA)(E/Z=99.5/0.5)和(Z)-6,10-二甲基十一碳-5-烯-2-酮(Z-DHGA)(Z/E=99/1)均被不对称氢化,以下列方式彼此分离:向2L高压釜中充入70g(0.353mol)特定异构体、700mL2,2,2-三氟乙醇和式(III-F)手性铱络合物在无水二氯甲烷(10g)中的溶液(570mg,0.356mmol,0.1mol%),所述式(III-F)手性铱络合物在所述式中*所指示的中心处具有表2给出的手性。将高压釜封闭并且施加压力为50巴的分子氢。将反应混合物在搅拌的同时加热至30℃,保持2小时。之后释放压力,除去溶剂。所形成的产物为(R)-6,10-二甲基十一烷-2-酮。所形成的异构体的转化率以及量如上述进行测定,结果在表2中给出。已合并两个单独/分开的不对称氢化的产物。E-DHGAZ-DHGAIr络合物的式III-FIII-F*处的手性Ir络合物的构型SR手性Ir络合物的量[摩尔%]0.10.1转化率[%]>99>99(R)-6,10-二甲基十一烷-2-酮[%]95.893.3(S)-6,10-二甲基十一烷-2-酮[%]4.26.7表2:E-DHGA和Z-DHGA的不对称氢化。在另一实验中,向高压釜中加入0.25mmol(E)-6,10-二甲基十一碳-5,9-二烯-2-酮(E-GA)或(E)-6,10-二甲基十一碳-5-烯-2-酮(E-DHGA)并分别加入0.5摩尔%、1摩尔%具有表2中给出的式的Ir络合物,和1.25ml纯(干燥)二氯甲烷。将高压釜封闭并且施加压力为50巴的分子氢。在搅拌下,将反应溶液在室温下保持14小时。之后释放压力,除去溶剂。为了测定转化率,在不进行任何进一步纯化的情况下,通过非手性气相色谱法分析粗产物。已使用使用上述方法测定了异构体的量。1234E-GAE-GAE-DHGAE-DHGAIr络合物的式III-FIII-CIII-DIII-C手性Ir络合物在*处的构型SSSS手性Ir络合物的量[摩尔%]0.50.511转化率[%]100100100100异构体-分布(6R)-6,10-二甲基十一烷-2-酮[%]96.5>9898.698.9(6S)-6,10-二甲基十一烷-2-酮[%]3.5<21.41.1表2a:E-GA或E-DHGA的不对称氢化。在两个另外的实验中,向高压釜中加入0.25mmol(E)-6,10-二甲基十一碳-5-烯-2-酮(E-DHGA)或(Z)-6,10-二甲基十一碳-5-烯-2-酮(Z-DHGA)和1摩尔%具有表2b或2c或2d中给出的式的Ir络合物和1.25ml表2b或2c中所示的纯(干燥)溶剂。将高压釜封闭并且施加压力为50巴的分子氢。在搅拌下,将反应溶液在室温下保持16小时。之后释放压力,除去溶剂。为了测定转化率,在不进行任何进一步纯化的情况下,通过非手性气相色谱法分析粗产物。已使用上述方法测定了异构体的量。5678E-DHGAE-DHGAE-DHGAE-DHGAIr络合物的式III-DIII-CIII-DIII-A'2手性Ir络合物在*处的构型RSRS手性Ir络合物的量[摩尔%]1111溶剂1TFEDCMDCMDCM转化率[%]>99>99>99>99异构体-分布(6R)-6,10-二甲基十一烷-2-酮[%]0.798.91.489.1(6S)-6,10-二甲基十一烷-2-酮[%]99.31.198.610.9表2b:通过不同Ir络合物进行的E-DHGA的不对称氢化。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2式(III-A')的手性Ir络合物:表2c:通过不同Ir络合物进行的E-DHGA的不对称氢化。1式的手性Ir络合物2TFE=2,2,2-三氟乙醇;DCM=二氯甲烷12131415Z-DHGAZ-DHGAZ-DHGAZ-DHGAIr络合物的式III-DIII-DIII-CIII-A'2手性Ir络合物在*处的构型RSSS手性Ir络合物的量[摩尔%]1111溶剂1TFEDCMDCMDCM转化率[%]>99>99>99>99异构体-分布(6R)-6,10-二甲基十一烷-2-酮[%]99.31.72.111.1(6S)-6,10-二甲基十一烷-2-酮[%]0.798.397.988.9表2d:通过不同Ir络合物进行的Z-DHGA的不对称氢化。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2式(III-A')的手性Ir络合物:实验E2a:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮的制备(步骤co)a)二甲基缩酮的制备将6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮(170.5mmol)添加至原甲酸三甲酯(50.8mL,49.2g,451mmol,2.65当量)中并冷却至5℃。在5min内添加MeOH(16mL)中的硫酸(96%,32.3mg,0.29mmol,0.2mol%)。随后,将反应加热回流(65℃IT)3h。冷却后,薄层色谱法(TLC)分析表明完全转化。添加NaOMe(0.24mL在MeOH中的25%溶液)来中和酸。将混合物真空浓缩,随后用己烷(50mL)稀释。滤出所形成的沉淀并浓缩滤液。通过蒸馏纯化粗产物,从而提供期望的二甲基缩酮。下文详细给出缩酮的表征。表2d(i):二甲基缩酮的制备。表征数据:(E)-10,10-二甲氧基-2,6-二甲基十一碳-2,6-二烯(E-GA-DM)1HNMR(300MHz,CDCl3):δ1.26(s,3H),1.58(s,3H),1.60(s,3H),叠加1.60-1.65(m,2H),1.66(brs,3H),1.92-2.09(m,6H),3.17(s,6H),5.02-5.14(m,2H)ppm。13CNMR(75MHz,CDCl3):δ15.9(1C),17.6(1C),20.8(1C),22.8(1C),25.6(1C),26.6(1C),36.4(1C),39.6(1C),47.9(2C),101.4(1C),123.8(1C),124.2(1C),131.2(1C),135.1(1C)ppm。MS(EI,m/z):240(M+,<1),225.3[(M-CH3)+,1],209.3[(M-CH3O)+,20],193.3(8),176.2(18),161.2(16),139.2(20),123.2(14),107.2(75),89.2(100),69.2(65),41.1(56)。IR(cm-1):2928(m),2857(w),2828(w),1670(w),1452(m),1376(s),1345(w),1302(w),1262(w),1222(w),1196(m),1172(m),1123(s),1102(s),1053(s),985(w),929(w),854(s),744(w),619(w)。(Z)-10,10-二甲氧基-2,6-二甲基十一碳-2,6-二烯(Z-GA-DM)1HNMR(300MHz,CDCl3):δ1.27(s,3H),1.56-1.65(m,5H),1.68(br.s,6H),1.96-2.09(m,6H),3.17(s,6H),5.11(t,J=7.2Hz,2H)ppm。13CNMR(75MHz,CDCl3):δ17.6(1C),20.9(1C),22.7(1C),23.3(1C),25.7(1C),26.6(1C),31.9(1C),36.7(1C),48.0(2C),101.4(1C),124.2(1C),124.6(1C),131.5(1C),135.4(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2943(m),2858(w),2828(w),1451(m),1376(m),1348(w),1301(w),1261(w),1197(m),1172(m),1153(w),1120(s),1098(m),1053(s),929(w),854(m),833(m),745(w),622(w)。(E)-2,2-二甲氧基-6,10-二甲基十一碳-5-烯(E-DHGA-DM)1HNMR(300MHz,CDCl3):δ0.83(d,J=6.6Hz,6H),1.02-1.13(m,2H),1.24(s,3H),1.27-1.39(m,2H),1.49(tqq,J=6.4,6.4,6,4Hz,1H),叠加1.53-1.63(m,2H),叠加1.56(s,3H),1.87-2.03(m,4H),3.13(s,6H),5.07(tq,J=7.0,1.4Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ16.1(1C),21.2(1C),23.0(2C),23.2(1C),26.0(1C),28.2(1C),36.9(1C),39.0(1C),40.2(1C),48.3(2C),101.8(1C),124.0(1C),135.9(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2953(s),2931(s),2870(m),2828(m),2108(w),1668(w),1460(m),1377(s),1367(m),1345(w),1301(w),1262(m),1221(m),1198(m),1172(s),1119(s),1100(s),1077(s),1053(s),967(w),927(w),854(w),796(w),739(w),620(w)。(Z)-2,2-二甲氧基-6,10-二甲基十一碳-5-烯(Z-DHGA-DM)1HNMR(300MHz,CDCl3):δ0.88(d,J=6.6Hz,6H),1.12-1.21(m,2H),1.28(s,3H),1.32-1.43(m,2H),1.53(dspt,J=6.6,6.6Hz,1H),1.57-1.66(m,2H),1.68(q,J=1.1Hz,3H),1.94-2.06(m,4H),3.18(s,6H),5.10(t,J=6.8Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ20.9(1C),22.6(2C),22.7(1C),23.3(1C),25.8(1C),27.9(1C),31.9(1C),36.8(1C),38.9(1C),48.0(2C),101.5(1C),124.3(1C),135.9(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2953(s),2870(w),2828(w),1461(w),1376(m),1301(w),1261(w),1205(m),1172(m),1119(m),1097(m),1074(m),1053(s),1022(w),927(w),854(m),738(w),621(w)。b)乙二醇缩酮的制备在氮气下,向反应容器充入二醇(112mL,125g,2.1mol)、对-甲苯磺酸一水合物(0.150g,0.5774mmol)和0.5mol(E)-6,10-二甲基十一碳-5-烯-2-酮或(Z)-6,10-二甲基十一碳-5-烯-2-酮中的任一者。将混合物在环境温度和减压(0.39毫巴)下搅拌5小时。在保持低压的同时,将温度缓慢升至40℃。在酮的转化率大于95%时,进一步升高温度,从而允许对乙二醇进行温和蒸馏,持续蒸馏直至达到大于99%的转化率。在室温下,通过三乙胺在庚烷中的溶液(2mL三乙胺/L庚烷)萃取产物。分离二醇相,用水中的NaHCO3溶液洗涤庚烷层。分离庚烷相,经无水Na2SO4干燥,真空过滤除去溶剂,得到粗缩酮。通过蒸馏进一步纯化缩酮。通过1H-NMR鉴定相应的缩酮。表2d(ii):乙二醇缩酮的制备。表征数据:(E)-2-(4,8-二甲基壬-3-烯-1-基)-2-甲基-1,3-二氧戊环(E-DHGA-en)1HNMR(300MHz,CDCl3)δ5.12(t,1H),3.95(m,4H),2.2-2(m,2H),1.94(t,2H),1.8-1.3(m,11H),1.2-1.0(m,2H),0.87(d,6H)ppm。(Z)-2-(4,8-二甲基壬-3-烯-1-基)-2-甲基-1,3-二氧戊环(Z-DHGA-en)1HNMR(300MHz,CDCl3)δ5.12(t,1H),3.94(m,4H),2.15-1.9(m,4H),1.7-1.45(m,6H),1.44-1.27(m,5H),1.23-1.08(m,2H),0.88(d,6H)ppm。c)新戊二醇缩酮的制备使表2d(iii)中示出的酮(90.7mmol)、2,2-二甲基-1,3-丙二醇(新戊二醇,32.4g,283mmol,3.4当量)和对甲苯磺酸一水合物(60mg,0.31mmol,0.3mol%)悬浮于甲苯(300mL)中。将反应加热至90℃,此时形成均匀溶液。随后,在75℃下,谨慎施加真空(先63毫巴,然后24毫巴)以缓慢蒸馏出甲苯(大约100mL,经4h)。4h后,薄层色谱法(TLC)分析表明酮完全转化。使反应冷却至室温,并且用庚烷(300mL)稀释,此时沉淀出过量新戊二醇。滤出沉淀(17.4g,湿重)。将滤液用Et3N(1mL)处理,随后用NaHCO3水溶液(2.4%w/w,2×300mL)洗涤,经MgSO4干燥并真空浓缩。通过蒸馏纯化粗产物,从而提供期望的新戊基缩酮。下文详细给出缩酮的表征。表2d(iii):新戊二醇缩酮的制备。表征数据:(E)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷(E-GA-neo)1HNMR(300MHz,CDCl3):δ0.92(s,3H),0.99(s,3H),1.37(s,3H),1.59(s,3H),1.61(s,3H),1.67(s,3H),1.68-1.75(m,2H),1.94-2.15(m,6H),AB信号(δA=3.46,δB=3.52,JAB=11.3Hz,4h),5.05-5.17(m,2H)ppm。13CNMR(75MHz,CDCl3):δ15.9(1C),17.6(1C),20.8(1C),22.0(1C),22.6(1C),22.7(1C),25.6(1C),26.7(1C),29.9(1C),37.3(1C),39.6(1C),70.3(2C),98.8(1C),124.1(1C),124.3(1C),131.2(1C),135.1(1C)ppm。MS(EI,m/z):280(M+,3),265[(M-CH3)+,14],176(21),129[(C7H13O2)+,100],69(63),43(43)。IR(cm-1):2954(m),2925(m),2858(m),2731(w),1720(w),1669(w),1473(w),1450(m),1394(m),1372(m),1349(w),1306(w),1271(w),1249(m),1211(m),1186(m),1123(s),1088(s),1043(m),1021(m),984(w),950(w),925(w),907(w),862(m),837(w),792(w),742(w),677(w),667(w)。(Z)-2-(4,8-二甲基壬-3,7-二烯-1-基)-2,5,5-三甲基-1,3-二噁烷(Z-GA-neo)1HNMR(300MHz,CDCl3):δ0.91(s,3H),0.97(s,3H),1.35(s,3H),1.60(s,3H),1.64-1.74(m,5H)叠加1.67(brs,3H),1.99-2.18(m,6H),AB信号(δA=3.44,δB=3.51,JAB=11.3Hz,4H),5.07-5.16(m,2H)ppm。13CNMR(75MHz,CDCl3):δ17.5(1C),20.9(1C),21.3(1C),21.9(1C),22.5(1C),22.6(1C),23.3(1C),25.7(1C),26.6(1C),29.9(1C),31.8(1C),37.5(1C),70.3(1C),98.7(1C),124.3(1C),124.9(1C),131.4(1C),135.2(1C)ppm。MS(EI,m/z):280(M+,3),265[(M-CH3)+,13],176(19),129[(C7H13O2)+,100],107(15),69(62),43(39)。IR(cm-1):2954(m),2927(m),2858(m),2729(w),1721(w),1671(w),1473(m),1450(m),1394(m),1374(m),1349(w),1315(w),1271(m),1249(m),1211(m),1187(m),1149(w),1120(s),1086(s),1043(m),1021(m),985(w),951(m),925(w),907(m),857(m),833(m),792(w),743(w),677(w),667(w)。(E)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷(E-DHGA-neo)1HNMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,6H),0.93(s,3H),1.00(s,3V),1.06-1.22(m,2H),1.31-1.43(m,2H)叠加1.38(s,3H),1.53(tqq,J=6.6,6.6,6.6Hz,1H),1.61(brs,3H),1.65-1.77(m,2H),1.94(t,J=7.5Hz,2H),2.05-2.17(m,2H),AB信号(δA=3.46,δB=3.54,JAB=11.4Hz,4H),5.13(tq,J=7.1,1.1Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ15.8(1C),20.9(1C),22.0(1C),22.59(1C),22.63(2C),22.7(1C),25.7(1C),27.9(1C),29.9(1C),37.3(1C),38.6(1C),39.9(1C),70.3(2C),98.8(1C),123.8(1C),135.6(1C)ppm。MS(EI,m/z):282(M+,5),267[(M-CH3)+,10),129(100),95(14),69(36),43(32)。IR(cm-1):2953(s),2929(m),2868(m),1720(w),1468(m),1394(m),1381(m),1368(m),1349(w),1306(w),1270(w),1250(m),1211(m),1187(w),1118(s),1087(s),1066(m),1044(m),1022(m),950(m),925(w),907(m),862(m),791(w),739(w),677(w),666(w)。(Z)-2-(4,8-二甲基壬-3-烯-1-基)-2,5,5-三甲基-1,3-二噁烷(Z-DHGA-neo)1HNMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,6H),0.93(s,3H),0.97(s,3H),1.10-1.20(m,2H),1.34-1.41(m,3H)叠加1.36(s,3H),1.53(tqq,J=6.6,6.6,6.6Hz,1H),1.64-1.75(m,2H)叠加1.67(q,J=1.5Hz,3H),1.95-2.15(m,4H),AB信号(δA=3.46,δB=3.51,JAB=11.1Hz,4H),5.12(brt,J=7.2Hz,1H)13CNMR(75MHz,CDCl3):δ21.1(1C),22.0(1C),22.61(3C),22.65(1C),23.4(1C),25.7(1C),27.9(1C),29.9(1C),31.9(1C),37.2(1C),38.8(1C),70.3(2C),98.8(1C),124.6(1C),135.8(1C)ppm。MS(EI,m/z):282(M+,6),267[(M-CH3)+,11),129(100),95(14),69(35),43(32)。IR(cm-1):2953(s),2867(m),1722(w),1468(m),1394(m),1368(m),1349(w),1306(w),1270(w),1250(m),1211(m),1189(w),1116(s),1086(s),1043(m),1022(m),951(m),925(w),907(m),856(m),792(w),739(w),677(w),667(w)。d)双(三氟乙基)缩酮的制备将250mL具有搅拌棒的三颈烧瓶在高真空下干燥(热风枪,250℃),然后使其冷却,用氩气冲洗并在氩气下充入1,1,1三氟乙醇(TFE)(40mL)。将烧瓶用冰浴冷却,同时在60min内滴加三甲基铝(2M,在庚烷中,20.0mL,40.0mmol,1.95当量),保持温度低于22℃。两相(TFE/庚烷)混合物在数分钟后再次变澄清,将其在室温下搅拌另外20min。在室温下,在5min内滴加20.7mmol如上所示制备的(E)-10,10-二甲氧基-2,6-二甲基十一碳-2,6-二烯(E-GA-DM)或(E)-2,2-二甲氧基-6,10-二甲基十一碳-5-烯(E-DHGA-DM)。1.5h后,GC分析表明起始物料完全转化。将反应用酒石酸钾钠在水中的半饱和溶液(100mL)淬灭,在室温下搅拌2h,最后用正己烷(200mL)稀释。将有机相分离,用正己烷(2×100mL)萃取,经MgSO4干燥并浓缩。粗产物通过柱色谱法(中性氧化铝,洗脱剂:正己烷)纯化。下文详细给出缩酮的表征。表2d(iv):双(三氟乙基)缩酮的制备。表征数据:(E)-2,6-二甲基-10,10-双(2,2,2-三氟乙氧基)十一碳-2,6-二烯(E-GA-tfe)1HNMR(300MHz,CDCl3):δ1.41(s,3H),1.62(brs,6H),1.67-1.76(m,2H),叠加1.69(q,J=0.9Hz,3H),1.93-2.15(m,6H),3.73-3.97(m,4H),5.02-5.18(m,2H)ppm。13CNMR(150MHz,CDCl3):δ15.9(1C),17.6(1C),21.3(1C),22.6(1C),25.7(1C),26.6(1C),36.9(1C),39.6(1C),59.3(q,JC,F=35.0Hz,2C),103.4(1C),124.0(q,JC,F=275.0Hz,2C),122.7(1C),124.1(1C),131.5(1C),136.2(1C)ppm。MS(EI,m/z):361[(M-CH3)+,1],276[(M-TFE)+,15],225[(CF3CH2O)2C-CH3)+,86],207(20),153(18),136(58),107(80),69(100),41(40)。IR(cm-1):2927(w),2859(w),1459(w),1419(w),1385(w),1281(s),1223(w),1156(s),1133(s),1081(s),971(s),889(m),860(w),845(w),678(w),663(w)。(E)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一碳-5-烯(E-DHGA-tfe)1HNMR(600MHz,CDCl3):δ0.88(d,J=6.8Hz,6H),1.11-1.17(m,2H),1.35-1.40(m,2H),1.41(s,3H),1.54(qqt,J=6.7,6.7,6.7Hz,1H),1.61(brs,3H),1.69-1.73(m,2H),1.95(t,J=7.7Hz,2H),2.03-2.09(m,2H),3.78-3.91(m,4H),5.09(tq,J=7.1,1.3Hz,1H)ppm。13CNMR(151MHz,CDCl3):δ14.1(1C),15.8(1C),21.3(1C),22.56(1C),22.61(1C),25.6(1C),27.9(1C),37.0(1C),38.6(1C),39.8(1C),59.2(q,JC,F=35.0Hz,2C),103.4(1C),124.0(q,JC,F=277.0Hz,2C),122.4(1C),136.7(1C)ppm。MS(EI,m/z):363[(M-CH3)+,1],278[(M-TFE)+,22],225[(CF3CH2O)2C-CH3)+,60],193(100),153(13),127(11),83(CF3CH2+,25),69(13),43(17)。IR(cm-1):2956(w),2933(w),2872(w),1462(w),1419(w),1385(w),1368(w),1281(s),1223(w),1156(s),1134(s),1081(s),971(s),889(m),860(w),845(w),679(w),663(m)。实验E2b:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的缩酮的不对称氢化向高压釜中充入0.5mmol表2e至2h中所示的6,10-二甲基十一碳-5,9-二烯-2-酮或6,10-二甲基十一碳-5-烯-2-酮的缩酮、4g表2e至2h中所示的溶剂以及表2e至2h中所示量的式(III-F)的手性铱络合物的溶液,所述式(III-F)的手性铱络合物在所述式中*所指示的中心处具有表2e至2h中所给出的手性。将高压釜封闭并且施加压力为30巴的分子氢。将反应混合物在室温下搅拌16小时。之后释放压力,除去溶剂。下文给出经氢化的缩酮的表征。表2e:E-6,10-二甲基十一碳-5,9-二烯-2-酮(E-GA)的不同缩酮的不对称氢化。条件:0.5mmol缩酮,4g溶剂,压力p(H2)=30巴,在室温下16h搅拌。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(R)表示6,10-二甲基十一烷-2-酮的相应缩酮的R-异构体,(S)表示其S-异构体3以缩酮水解后的酮测定表2f:Z-6,10-二甲基十一碳-5,9-二烯-2-酮(Z-GA)的不同缩酮的不对称氢化。条件:0.5mmol缩酮,4g溶剂,压力p(H2)=30巴,在室温下16h搅拌。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(R)表示6,10-二甲基十一烷-2-酮的相应缩酮的R-异构体,(S)表示其S-异构体3以缩酮水解后的酮测定表2g:E-DHGA的不同缩酮的不对称氢化。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(R)表示6,10-二甲基十一烷-2-酮的相应缩酮的R-异构体,(S)表示其S-异构体3以缩酮水解后的酮测定表2h:Z-DHGA的不同缩酮的不对称氢化。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(R)表示6,10-二甲基十一烷-2-酮的相应缩酮的R-异构体,(S)表示其S-异构体3以缩酮水解后的酮测定表征数据:(R)-2,2-二甲氧基-6,10-二甲基十一烷(R-THGA-DM)1HNMR(300MHz,CDCl3):δ0.848(d,J=6.6Hz,3H)叠加0.852(d,J=6.6Hz,6H),1.01-1.41(m,11H)叠加1.25(s,3H),1.44-1.61(m,3H),3.16(s,6H)ppm。13CNMR(75MHz,CDCl3):δ14.1(1C),19.6(1C),20.9(1C),21.7(1C),22.6(1C),22.7(1C),24.8(1C),27.9(1C),32.7(1C),36.8(1C),37.2(1C),37.4(1C),39.3(1C),47.9(1C),101.7(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2951(s),2927(m),2870(m),2828(m),1723(w),1462(m),1377(m),1309(w),1256(m),1215(m),1194(m),1172(m),1111(m),1089(m),1053(s),972(w),934(w),920(w),855(m),815(m),736(w),618(w)。(R)-2-(4,8-二甲基壬基)-2,5,5-三甲基-1,3-二噁烷(R-THGA-neo)1HNMR(300MHz,CDCl3):δ0.87(d,J=6.6Hz,9H),0.91(s,3H),1.01(s,3H),1.04-1.61(m,12H)叠加1.36(s,3H),1.61-1.74(m,2H),AB信号(δA=3.44,δB=3.54,JAB=11.7Hz,4H)ppm。13CNMR(75MHz,CDCl3):δ19.7(1C),20.4(1C),21.0(1C),22.56(1C),22.61(1C),22.71(1C),22.77(1C),24.8(1C),28.0(1C),30.0(1C),32.8(1C),37.3(1C),37.4(1C),38.2(1C),39.3(1C),70.3(2C),99.1(1C)ppm。MS(EI,m/z):269[(M-CH3)+,65),199(8),129(100),109(8),69(32),55(10),43(25)。IR(cm-1):2953(s),2925(s),2868(m),1722(w),1464(m),1394(m),1371(m),1316(w),1258(m),1212(m),1161(m),1141(m),1111(s),1095(s),1043(m),1020(m),951(m),925(m),907(m)870(m),855(m),801(m),792(m),737(m),677(w),667(w)。(R)-6,10-二甲基-2,2-双(2,2,2-三氟乙氧基)十一烷(R-THGA-tfe)1HNMR(300MHz,CDCl3):δ0.88(d,J=6.6Hz,6H),0.87(d,J=6.4Hz,3H),1.03-1.23(m,5H),1.39(s,3H),1.38-1.40(m,6H),1.46-1.71(m,3H),3.73-3.94(m,4H)。13CNMR(75MHz,CDCl3):δ19.5(1C),21.39(1C),21.47(1C),22.58(1C),22.68(1C),24.7(1C),28.0(1C),32.6(1C),37.0(1C),37.19(1C),37.23(1C),39.3(1C),59.2(q,2JC,F=32.5Hz,2C),103.6(1C),124.1(q,1JC,F=279.0Hz,2C)。MS(EI,m/z):365[(M-CH3)+,1],281(2),225[(CF3CH2O)2C-CH3)+,100],153(8),140(6),83(CF3CH2+,6),43(7)。IR(cm-1):2955(w),2929(w),2872(w),1463(w),1419(w),1385(w),1281(s),1216(w),1156(s),1122(m),1082(s),972(m),892(m),861(w),737(w),679(w),663(m)。实验E2c:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的经氢化的缩酮的水解在(6R,10R)-6,10,14-三甲基十五烷-2-酮的缩酮不对称氢化后,获得的经氢化的缩酮水解成酮并且分别生成(R)-6,10-二甲基十一烷-2-酮或(S)-6,10-二甲基十一烷-2-酮。方法1–来自二氯甲烷中不对称氢化反应的新戊基缩酮、二甲基缩酮将来自不对称氢化反应的反应混合物的样品(1-2ml)用等体积的1M盐酸水溶液在室温下搅拌1小时。添加二氯甲烷(2ml),分离各层。将水层用二氯甲烷(2ml)洗涤两次。将合并的有机层减压蒸发,得到呈无色至浅黄色油状物的酮。然后分析粗酮的纯度和异构体比率。方法2–来自三氟乙醇中不对称氢化反应的乙二醇缩酮、双(三氟乙醇)缩酮和二甲基缩酮将来自不对称氢化反应的反应混合物的样品(1-2ml)用9:1:0.2(按体积计)的甲醇:水:三氟乙酸的0.5ml溶液在40℃下搅拌1小时。添加二氯甲烷(2ml)和水(2ml),分离各层。将水层用二氯甲烷(2ml)洗涤两次。将合并的有机层减压蒸发,得到呈无色至浅黄色油状物的酮。然后分析粗酮的纯度和异构体比率。实验E2d:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮的酮和缩酮的不对称氢化在氮气下向高压釜容器充入在*标记的手性中心处具有R构型的式(III-F)的手性铱络合物、表2h或2i中所示的酮或缩酮(浓度)、表2h或2i中所示的溶剂)。将反应容器封闭,并用分子氢加压至表2h或2i中所示的压力(pH2)。在氢气下,将反应混合物在室温下搅拌时间(t),如表2h或2i中所示。然后释放压力,测定经完全氢化的产物的测定收率和立体异构体分布。就缩酮而言,已在如实验E2c中所示对缩酮进行水解后测定了测定收率和立体异构体分布。催化剂负载(S/C)被定义为mmol酮或缩酮("底物")/mmol手性铱络合物。3233待氢化的酮E-DHGA待氢化的缩酮E-DHGA-en浓度1[mol/L]1.00.9pH2[巴]5050t[h]2020S/C10'00010'000溶剂TFETFE测定收率[面积%]197异构体-分布3,4(R)[%]n.d.22.2(S)[%]n.d.297.8表2hE-DHGA和E-DHGA-en的氢化。缩酮化的影响。1浓度=mol酮或缩酮/L溶剂2n.d.=未测定(由于低测定收率)3(R)表示6,10-二甲基十一烷-2-酮的乙二醇缩酮的R-异构体,(S)表示其S-异构体4以缩酮水解后的酮测定表2iZ-DHGA、Z-DHGA-en和Z-DHGA-neo的氢化。缩酮化的影响。1浓度=mol酮或缩酮/L溶剂(DCM=二氯甲烷)2n.d.=未测定(由于低测定收率)3(R)表示6,10-二甲基十一烷-2-酮的乙二醇缩酮的R-异构体,(S)表示其S-异构体4以缩酮水解后的酮测定实验E2e:6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮或其缩酮在添加剂的存在下的不对称氢化按照实验E2d进行不对称氢化,不同之处在于氢化使用添加剂。表2j、2k示出用于氢化缩酮的添加剂和用量,表2l和2m示出用于氢化酮的添加剂和用量。添加剂的制备-MAO/TFE:将1.6MMAO(MAO:甲基铝氧烷)在甲苯(0.64mL)中的溶液用2,2,2-三氟乙醇(TFE)(3.1mmol)淬灭,从而产生少量过量的游离TFE。-EAO/TFE:将10重量%EAO(EAO:乙基铝氧烷)在甲苯(1mmol)中的溶液用TFE(3.2mmol)淬灭,从而产生少量过量的游离TFE。-TMA/TFE:将2MTMA(TMA:三甲基铝(Al(CH3)3))在庚烷(1mmol)中的溶液用TFE(3.1mmol)淬灭,从而产生少量过量的游离TFE。-TEA/TFE:将2MTEA(TEA:三乙基铝(Al(CH2CH3)3))在庚烷(1mmol)中的溶液用TFE(3.1mmol)淬灭,从而产生少量过量的游离TFE。-TMA/BHT/TFE:将2MTMA在庚烷(1mmol)中的溶液先后用2,6-二-叔丁基-4-甲基苯酚(BHT)(2mmol)和TFE(3.1mmol)淬灭,从而产生少量过量的游离TFE。-Ti(OCH2CF3)4:将原钛酸四异丙酯(8.1mmol)在50℃下溶解于2,2,2-三氟乙醇中。除去溶剂,得到呈白色残留物的Ti(OCH2CF3)4,将其分离,鉴定为Ti(OCH2CF3)4。这些添加剂为新鲜制备的并且在室温下作为均匀混合物使用,或通过加热至介于50℃和70℃之间的温度而均匀。添加剂原钛酸四异丙酯(Ti(OiPr)4)、硼酸三异丙酯(B(OiPr)3)、四[3,5-双(三氟甲基)苯基]硼酸钠(NaBArF)和三乙基硼烷(TEB)(1M己烷溶液)可商购获得并且按收到的原样使用。表2jE-DHGA-en在50巴的分子氢(pH2)压力下氢化并且在20小时内在室温下进行搅拌。添加剂的影响。1浓度=mol缩酮/L溶剂2相对于E-DHGA-en的摩尔量3(R)表示6,10-二甲基十一烷-2-酮的乙二醇缩酮的R-异构体,(S)表示其S-异构体4以缩酮水解后的酮测定。表2kZ-DHGA的不同缩酮在50巴的分子氢(pH2)压力下氢化并且在20小时内在室温下进行搅拌。添加剂的影响。1浓度=mol缩酮/L溶剂2相对于Z-DHGA的缩酮的摩尔量3TFE=2,2,2-三氟乙醇;DCM=二氯甲烷4TMA通过加入到溶剂TFE中来被淬灭5(R)表示6,10-二甲基十一烷-2-酮的相应缩酮的R-异构体,(S)表示其S-异构体6以缩酮水解后的酮测定表2lE-DHGA在50巴的分子氢(pH2)压力下氢化并且在20小时内在室温下进行搅拌。添加剂的影响。1浓度=mol酮/L溶剂2相对于E-DHGA的摩尔量。3n.d.=未测定(由于低测定收率)5455待氢化的酮Z-DHGAZ-DHGA浓度1[mol/L]1.00.8S/C5'0005'000溶剂DCMDCM添加剂-TMA/TFE添加剂浓度[摩尔%]2-5测定收率[面积%]140(R)-6,10-二甲基十一烷-2-酮[%]n.d.398.3(S)-6,10-二甲基十一烷-2-酮[%]n.d.31.7表2mZ-DHGA在50巴的分子氢(pH2)压力下氢化并且在20小时内在室温下进行搅拌。添加剂的影响。1浓度=mol酮/L溶剂2相对于Z-DHGA的摩尔量3n.d.=未测定(由于低测定收率)。化学转化步骤(步骤d)实验E3a:(R)-6,10-二甲基十一烷-2-酮的乙烯化(步骤d1’)将配备有顶置式搅拌器、温度计、冷凝器和氩气入口的100mL干燥四颈烧瓶抽真空,并且用氩气吹扫。在室温下添加乙烯基氯化镁(23.63mL1.6MTHF溶液,37.8mmol,1.56当量)。在20min内缓慢添加(R)-6,10-二甲基十一烷-2-酮(5.01g,24.24mmol,96.0%,1.0当量)在无水THF(20mL)中的溶液。通过用冰浴冷却使放热反应维持在介于25℃和30℃之间的内部温度。结束添加后,将反应在室温下搅拌1h。小心添加饱和NH4Cl溶液(10mL)以淬灭过量的格氏试剂。添加戊烷(150mL)、水(150mL)和盐水(150mL)。将有机相用盐水(2×150mL)萃取,并将水相用戊烷(2×150mL)反萃取。将合并的有机相干燥(MgSO4)并真空浓缩,得到无色油状物(5.42g)。在Kugelrohr设备中通过真空蒸馏来纯化粗产物。将主要馏分在143℃/3.8-2毫巴下进行蒸馏,得到呈无色油状物的(7R)-3,7,11-三甲基十二碳-1-烯-3-醇,纯度为94.0%(5.15g,21.38mmol,88%收率)。实验E3b:(R)-6,10-二甲基十一烷-2-酮的乙炔化(步骤d1)将(R)-6,10-二甲基十一烷-2-酮(340g,1.70mol,1.0当量,99.0%)添加至配备有恒温器、计量泵、乙炔入口和氨气入口的高压釜中。将反应器密封,抽真空,用氮气冲洗,然后冷却至15℃。将氨气(632g,37.2mol,22.0当量,99.8%)冷凝到反应器中并冷却至15℃,从而产生8-9巴的压力。引入乙炔直至达到12巴的压力,然后在15℃下定量添加KOH(在水中45重量%,6.6g,52.9mmol,3.1mol%)。反应过程通过GC来监测。90min后,将反应混合物用乙酸中和,随后在25℃下对反应器进行排气。将反应混合物洗涤,真空浓缩并通过真空蒸馏来纯化,得到325g(7R)-3,7,11-三甲基十二碳-1-炔-3-醇,纯度为95%(81%收率)。实验E3c:(7R)-3,7,11-三甲基十二碳-1-炔-3-醇的氢化(步骤d2)将(7R)-3,7,11-三甲基十二碳-1-炔-3-醇(910g,4.06mol,1.0当量,95%),林德拉催化剂(850mg)加入高压釜中。将反应器密封,抽真空,用氮气冲洗,随后加热至45℃。将反应器再次抽真空,用氢气冲洗,然后加压至2巴。将反应搅在45℃下拌大约2-3小时,直至已消耗计算量的分子氢。过滤后,获得884g(7R)-3,7,11-三甲基十二碳-1-烯-3-醇,纯度为91.5%(88%收率)。实验E4:(7R)-3,7,11-三甲基十二碳-1-烯-3-醇与2-甲氧基丙-1-烯的反应(步骤d3)将(7R)-3,7,11-三甲基十二碳-1-烯-3-醇(640g,2.59mol,1.0当量,91.5%)、H3PO4(水中20重量%,4.5g,8.2mmol,0.32mol%)和异丙烯基甲基醚(600g,8.17mol,3.2当量,98.0%)加入配备有恒温器、顶置式搅拌器和1.1m蒸馏柱(Sulzer填料)的2L高压釜中。将反应器密封,抽真空,用氮气冲洗,随后加热至80℃。缓慢排放所积聚的压力。然后在1h内使内部温度逐渐升至160℃。将反应物在160℃下搅拌3h。将反应混合物冷却至30℃,真空浓缩,然后用水和NaHCO3溶液洗涤。通过蒸馏(夹套温度150℃,1毫巴)除去低沸点物质,得到690g呈残留物的(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的混合物,纯度为85%(85%收率)。混合物经GC分析为49%E-异构体和36%Z-异构体的混合物。实验E5:(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的E/Z异构体混合物的分离(步骤e)使用由蒸馏器(容积:9升)、降膜蒸发器、精馏柱(70mm内径,高度5m)组成的分离设备对(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的1.94kg混合物(36%(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮和49%(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮)进行分馏。柱配备有非常有效的规整填料(Sulzer)。精馏过程在大约2毫巴的顶部压力、95℃至122℃的柱顶部温度以及165℃的蒸馏器底部温度下进行。回流比率调节至20。分馏馏出液料流,得到包含(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的馏分(Z-异构体的含量=97%)。最后,发现(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮留在蒸馏器中(E-异构体的含量=94%)。通过分馏对两种异构体进行进一步纯化,得到E-异构体和Z-异构体两者的馏分,每一者的纯度为99.5%)实验E6:(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的不对称氢化(步骤f)两种异构体(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮)(R,E-THFA)(E/Z=99.5/0.5,R/S=92/8)和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮(R,Z-THFA)(Z/E=99.5/0.5,R/S=92/8)均被不对称氢化,以下列方式彼此分离:向125mL高压釜中充入7.0g(26mmol)特定异构体、50mL2,2,2-三氟乙醇和式(III-F)手性铱络合物在无水二氯甲烷(4g)中的溶液(42mg,0.026mmol,0.1mol%),所述手性铱络合物在所述式中*所指示的中心处具有表3给出的手性。将高压釜封闭并且施加压力为50巴的分子氢。将反应混合物在搅拌的同时加热至30℃,保持16小时。之后释放压力,除去溶剂。所形成的产物为(6R,10R)-6,10,14-三甲基十五烷-2-酮。所形成的异构体的转化率以及量在表3中给出。已合并两个单独/分开的不对称氢化的产物。在另一实验中,向高压釜中加入0.25mmol(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮)(R,E-THFA)和1摩尔%式(III-D)的Ir络合物和1.25ml纯(干燥)二氯甲烷。将高压釜封闭并且施加压力为50巴的分子氢。在搅拌下,将反应溶液在室温下保持14小时。之后释放压力,除去溶剂。为了测定转化率,在不进行任何进一步纯化的情况下,通过非手性气相色谱法分析粗产物。异构体的量已使用以上方法进行了测定并且在表3中给出。在另一实验中,向高压釜中加入0.5mmol(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮)(R,E-THFA)和1摩尔%具有如表3'中所示的式的Ir络合物和4g如表3'中所示的溶剂。将高压釜封闭并且施加压力为30巴的分子氢。在搅拌下,将反应溶液在室温下保持16小时。之后释放压力,除去溶剂。为了测定转化率,在不进行任何进一步纯化的情况下,通过非手性气相色谱法分析粗产物。异构体的量已使用以上方法进行了测定并且在表3'中给出。表3:R,E-THFA和R,Z-THFA的不对称氢化。*:(6R,10S)和(6S,10S)异构体合计测定。表3':通过不同Ir络合物进行的R,E-THFA和R,Z-THFA的不对称氢化。1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2式(III-A')的手性Ir络合物:实验E6a:(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮的制备(步骤fo)类似于以上针对6,10-二甲基十一碳-5-烯-2-酮或6,10-二甲基十一碳-5,9-二烯-2-酮所述的实验E2a,已分别获得(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的二甲基缩酮、新戊二醇缩酮或双(三氟乙基)缩酮。表3a:6,10,14-三甲基十五碳-5-烯-2-酮的二甲基和新戊二醇缩酮的制备表征数据:(R,E)-2,2-二甲氧基-6,10,14-三甲基十五碳-5-烯(R-E-THFA-DM)1HNMR(300MHz,CDCl3):δ0.84(d,J=6.6Hz,3H),叠加0.86(d,J=6.6Hz,6H),0.99-1.44(m,11H),叠加1.28(s.3H),1.52(tqq,J=6.6,6.6,6.6Hz,1H),1.60(s,3H),1.60-1.66(m,2H),1.90-2.05(m,4H),3.18(s,6H),5.10(tq,J=7.1,1.1Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ16.3(1C),20.1(1C),21.3(1C),23.0(1C),23.1(1C),23.2(1C),25.2(1C),25.7(1C),28.4(1C),33.1(1C),36.9(1C),37.1(1C),37.7(1C),39.8(1C),40.3(1C),48.4(2C),101.9(1C),124.0(1C),136.0(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2952(m),2927(s),2869(m),2828(w),1461(m),1377(m),1301(w),1262(m),1222(m),1197(m),1172(m),1120(m),1101(m),1076(m),1054(s),930(w),854(m),737(w),620(w)。(R,Z)-2,2-二甲氧基-6,10,14-三甲基十五碳-5-烯(R-Z-THFA-DM)1HNMR(300MHz,CDCl3):δ0.85(d,J=6.4Hz,3H),叠加0.87(d,J=6.4Hz,6H),1.01-1.27(m,7H),1.28(s,3H),1.29-1.44(m,4H),1.53(dqq,J=6.5,6.5Hz,6.5Hz,1H),1.58-1.66(m,2H),1.68(q,J=1.1Hz,3H),1.91-2.08(m,4H),3.18(s,6H),5.11(t,J=6.8Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ)19.7(1C),20.9(1C),22.60(1C),22.69(1C),22.71(1C),23.4(1C),24.8(1C),25.5(1C),28.0(1C),32.0(1C),32.7(1C),36.8(1C),37.0(1C),37.3(1C),39.3(1C),48.0(2C),101.5(1C),124.3(1C),135.9(1C)ppm。MS(EI,m/z):由于在柱上降解而未获得GC-MS。IR(cm-1):2952(m),2927(m),2869(m),2828(w),1462(m),1376(m),1301(w),1261(w),1197(w),1172(m),1119(m),1098(m),1074(m),1054(s),1022(w),854(m),736(w),622(w)。(R,E)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷(R-E-THFA-neo)1HNMR(300MHz,CDCl3):δ0.84(d,J=6.4Hz,3H),0.86(d,J=6.6Hz,6H),0.92(s,3h),0.99(s,3H),叠加0.97-1.44(m,11H),叠加1.37(s,3H),1.52(qqt,J=6.9,6.9,6.9Hz,1H),1.60(s,3H),1.67-1.76(m,2H),1.93(t,J=7.4Hz,2H),2.03-2.18(m,2H),AB信号(δA=3.45,δB=3.52,JAB=11.4Hz,4H),5.12(tq,J=7.2,1.0Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ15.8(1C),19.7(1C),20.9(1C),22.0(1C),22.6(2C),22.7(2C),24.8(1C),25.3(1C),27.9(1C),29.9(1C),32.6(1C),36.6(1C),37.3(1C),37.4(1C),39.3(1C),39.9(1C),70.3(2C),98.8(1C),123.8(1C),135.5(1C)ppm。MS(EI,m/z):352(M+,4),337[(M-CH3)+,8),265(6),129(100),95(10),69(25),43(25)。IR(cm-1):2953(s),2926(s),2867(m),1462(m),1394(w),1369(m),1270(w),1249(m),1211(m),1187(w),1119(s),1088(s),1043(m),1021(m),951(w),925(w),907(w),862(m),791(w),738(w),678(w)。(R,Z)-2,5,5-三甲基-2-(4,8,12-三甲基十三碳-3-烯-1-基)-1,3-二噁烷(R-Z-THFA-neo)1HNMR(300MHz,CDCl3):δ0.84(d,J=6.4Hz,3H),叠加0.86(d,J=6.6Hz,6H),0.93(s,3H),0.97(s,3H),1.00-1.42(m,11H),叠加1.36(s,3H),1.52(qqt,J=6.7,6.7,6.7Hz,1H),1.63-1.76(m,2H),1.67(s,3H),1.94-2.15(m,4H),AB信号(δA=3.45,δB=3.51,JAB=11.1Hz,4H),5.12(t,J=7.1Hz,1H)ppm。13CNMR(75MHz,CDCl3):δ19.6(1C),21.1(1C),21.9(1C),22.60(2C),22.67(2C),22.69(1C),23.4(1C),24.8(1C),25.4(1C),27.9(1C),29.9(1C),32.0(1C),32.7(1C),36.9(1C),37.3(1C),39.3(1C),70.3(2C),98.8(1C),124.6(1C),135.7(1C)ppm。MS(EI,m/z):352(M+,3),337[(M-CH3)+,9),265(6),129(100),95(10),69(24),43(25)。IR(cm-1):2953(s),2926(s),2860(m),1463(m),1394(w),1371(m),1270(w),1250(w),1211(m),1188(w),1117(s),1086(s),1043(m),1022(w),951(w),925(w),907(w),855(m),792(w),737(w),667(w)。实验E6b:(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮的不对称氢化向高压釜中充入0.5mmol表3b和3c中所示的(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的缩酮、4g表3b和3c中所示的溶剂以及表3b和3c中所示量的式(III-F)手性铱络合物的溶液,所述式(III-F)手性铱络合物在所述式中*所指示的中心处具有表3b和3c中所给出的手性。将高压釜封闭并且施加压力为30巴的分子氢。将反应混合物在室温下搅拌16小时。之后释放压力,除去溶剂。使用在*标记的手性中心处具有S构型的式(III-F)的Ir络合物氢化(R,E)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五碳-5-烯,生成(6R,10R)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷。下文给出经氢化的缩酮的表征。表3b:产生(6R,10R)-6,10,14-三甲基十五烷-2-酮的(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮的不同缩酮的不对称氢化。条件:0.5mmol缩酮,4g溶剂,压力p(H2)=30巴,在室温下16h搅拌1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(SS)表示6,10,14-三甲基十五烷-2-酮的对应的缩酮的(6S,10S)-异构体,(RR)表示其(6R,10R)-异构体,(SR)表示其(6S,10R)-异构体,(RS)表示其(6R,10S)-异构体3以缩酮水解后的酮测定表3c:(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的不同缩酮的不对称氢化产生(6R,10R)-6,10,14-三甲基十五烷-2-酮。条件:0.5mmol缩酮,4g溶剂,压力p(H2)=30巴,在室温下16h搅拌1TFE=2,2,2-三氟乙醇;DCM=二氯甲烷2(SS)表示6,10,14-三甲基十五烷-2-酮的对应的缩酮的(6S,10S)-异构体,(RR)表示其(6R,10R)-异构体,(SR)表示其(6S,10R)-异构体,(RS)表示其(6R,10S)-异构体3以缩酮水解后的酮测定。表征数据:(6R,10R)-2,2-二甲氧基-6,10,14-三甲基十五烷(RR18-DM)1HNMR(300MHz,CDCl3):δ0.83-0.89(m,12H),0.98-1.45(m,21H),1.46-1.65(m,3H),3.18(s,6H)。13CNMR(75MHz,CDCl3):δ19.68(1C),19.73(1C),21.0(1C),21.7(1C),22.6(1C),22.7(1C),24.5(1C),24.8(1C),28.0(1C),32.72(1C),32.78(1C),36.8(1C),37.28(1C),37.33(1C),37.36(1C),37.41(1C),39.4(1C),48.0(2C),101.7(1C)ppm。IR(cm-1):2951(s),2926(s),2869(s),2828(m),1734(w),1723(w),1216(w),1463(s),1377(s),1308(w),1255(m),1215(m),1172(s),1105(s),1090(s),1054(s),971(w),933(w),860(s),815(m),736(w)618(w)。2,5,5-三甲基-2-((4R,8R)-4,8,12-三甲基十三基)-1,3-二噁烷(RR18-neo)1HNMR(300MHz,CDCl3):δ0.78-0.95(m,15H),0.95-1.61(m,19H),叠加1.01(s,3H),1.36(s,3H),1.63-1.74(m,2H),AB信号(δA=3.44,δB=3.55,JAB=11.7Hz,4H)ppm。13CNMR(75MHz,CDCl3):δ19.72(1C),19.74(1C),20.4(1C),20.9(1C),22.56(1C),22.62(1C),22.72(1C),22.77(1C),24.5(1C),24.8(1C),28.0(1C),30.0(1C),32.8(1C),32.8(1C),37.28(1C),37.35(1C),37.42(2C),38.2(1C),39.4(1C),70.3(2C),99.1(1C)ppm。MS(EI,m/z):339[(M-CH3)+,83],269(5),129(100),69(21),43(18)。IR(cm-1):2952(s),2925(s),2867(m),1463(m),1394(m),1372(m),1258(m),1211(m),1189(w),1141(w),1100(s),1043(m),1020(m),951(w),925(w),907(m),858(m),792(w),737(w),677(w)。(6R,10R)-6,10,14-三甲基-2,2-双(2,2,2-三氟乙氧基)十五烷(RR18-tfe)1HNMR(600MHz,CDCl3):δ0.86(d,J=6.6Hz,3H),0.879(d,J=6.6Hz,3H),0.882(d,J=6.6Hz,3H),0.884(d,J=6.6Hz,3H),1.03-1.46(m,18H),叠加1.40(s,3H),1.54(qqt,J=6.6,6.6,6.6Hz,1H),1.60-1.70(m,2H),3.77-3.90(m,4H)ppm。13CNMR(151MHz,CDCl3):δ19.6(1C),19.7(1C),21.4(1C),21.5(1C),22.6(1C),22.7(1C),24.5(1C),24.8(1C),28.0(1C),32.6(1C),32.8(1C),37.0(1C),37.24(1C),37.30(1C),37.34(1C),37.43(1C),39.4(1C),59.2(q,2JC,F=35.0Hz,2C),103.6(1C),124.0(q,1JC,F=277.0Hz,2C)ppm。MS(EI,m/z):435[(M-CH3)+,1],351(1),250(1),225[(CF3CH2O)2C-CH3)+,100],153(7),140(5),83(CF3CH2+,3),43(6)。IR(cm-1):2954(m),2927(m),2871(w),1463(w),1419(w),1384(w),1281(s),1215(w),1157(s),1123(m),1082(s),972(s),892(m),861(w),737(w),679(w),663(m)。实验E6c:(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的经氢化的缩酮的水解(R,E)-6,10,14-三甲基十五碳-5-烯-2-酮和(R,Z)-6,10,14-三甲基十五碳-5-烯-2-酮的相应缩酮不对称氢化后,所获得的经氢化的缩酮被水解成酮并生成(6R,10R)-6,10,14-三甲基十五烷-2-酮。经氢化的缩酮已如实验E2c中所述被水解。(R,R)-异植醇的形成实验E6-I:(6R,10R)-6,10,14-三甲基十五烷-2-酮的乙炔化(步骤g)将(6R,10R)-6,10,14-三甲基十五烷-2-酮(35.0g,129mmol,1.0当量,98.8%)添加至配备有恒温器、计量泵、乙炔入口和氨气入口的高压釜中。将反应器密封,抽真空,然后用氮气冲洗,并冷却至15℃。将氨气(715g,45.0mol,326当量,99.8%)冷凝到反应器中并冷却至15℃,从而产生8-9巴的压力。引入乙炔直至达到12巴,然后在15℃下定量添加KOH(40重量%水中,5.0g,35.6mmol,28mol%)。反应过程通过GC来监测。在达到期望的转化率时(大约2h后),将反应混合物用乙酸中和,随后在25℃下对反应器进行排气。将反应混合物洗涤,真空浓缩并通过真空蒸馏来纯化,得到26.9g(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇,纯度为98.8面积%(70%收率)。实验E6-II:在林德拉催化剂的存在下氢化(6R,10R)-6,10,14-三甲基十五烷-2-酮(步骤h)将溶解于庚烷(40g)中的(7R,11R)-3,7,11,15-四甲基十六碳-1-炔-3-醇(10g,33.4mmol,98.4%纯度)和林德拉催化剂(850mg)加入高压釜中。将反应器密封,用氮气冲洗,随后加热至85℃。当达到期望的温度时,用2巴氢气对反应进行加压。将反应在此温度下搅拌大约22小时,直至消耗期望的量的氢气。过滤后,将粗产物与第二反应批料合并。将11.9g粗料通过蒸馏来纯化,得到11.1g(R,R)-异植醇(经GC测定97.6%纯度,88%总收率)。实验E6-III:(6R,10R)-6,10,14-三甲基十五烷-2-酮的乙烯化(步骤h’)将配备有顶置式搅拌器、温度计、冷凝器和氩气入口的100mL干燥四颈烧瓶抽真空,并且用氩气吹扫。在室温下添加乙烯基氯化镁(18.3mL1.6MTHF溶液,29.0mmol,1.59当量)。在25min内缓慢添加(6R,10R)-6,10,14-三甲基十五烷-2-酮(5.00g,18.3mmol,98.2%,1.0当量)在无水THF(20mL)中的溶液。通过用冰浴冷却使放热反应维持在介于25℃和30℃之间的内部温度。结束添加后,将反应在室温下搅拌1h。小心添加饱和NH4Cl溶液(10mL)以淬灭过量的格氏试剂。添加戊烷(150mL)、水(150mL)和盐水(150mL)。将有机相用盐水(2×150mL)萃取,并将水相用戊烷(2×150mL)反萃取。将合并的有机相干燥(MgSO4)并真空浓缩,得到无色油状物(5.58g)。在Kugelrohr设备中通过真空蒸馏来纯化粗产物。将主要馏分在143℃/3.5×10-2毫巴下进行蒸馏,得到呈无色油状物的(R,R)-异植醇(=(7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇),纯度为99.3%(5.271g,96%收率)。实验E7:(2-双型)-α-生育酚的形成(步骤m)根据WO2005/121115A1中公开的程序,(R,R)-异植醇(=(7R,11R)-3,7,11,15-四甲基十六碳-1-烯-3-醇)与2,3,5-三甲基苯-1,4-二醇(=2,3,5-三甲基氢醌)在缩合催化剂的存在下缩合成(2-双型)-α-生育酚。实验E8:(2R,4’R,8’R)-α-生育酚的形成(步骤n)(2-双型)-α-生育酚使用手性相通过色谱分离来分离。制备型色谱法产生(2R,4’R,8’R)-α-生育酚和(2S,4’R,8’R)-α-生育酚:实验E7的(2-双型)-α-生育酚通过HPLC(柱:DaicelOD-H,250mm×4.6mm;洗脱剂:正庚烷中的0.5%乙醇;流量1ml/min;检测220nm,2μl进样)来分析。图7b)示出此色谱图(保留时间分别为7.2min和8.2min,50.2:49.2)。对140mg(2-双型)-α-生育酚在庚烷中的溶液进行进样,通过制备型HPLC分离对保留时间最多为13.4min(1)(50.1%)和15.0min(2)(49.9%)的两个峰进行分离。图7a)示出制备型HPLC分离的色谱图。在蒸干和溶解后,在分析柱(DaicelOD-H,250mm×4.6mm;洗脱剂:正庚烷中的0.5%乙醇;流速1ml/min;检测220nm,2μl进样)上对两个收集的馏分进行了重新分析。图7c)和图7d)分别示出第一馏分和第二馏分的色谱图。所述馏分中两种异构体(保留时间分别为7.2min和8.2min)的异构体比率分别为99.5:0.5(图7c))和0.8:99.2(图7d)。因此,通过制备型色谱法几乎完全分离了两种异构体。所述异构体已鉴定为(2R,4’R,8’R)-α-生育酚(保留时间7.2min)和(2S,4’R,8’R)-α-生育酚(保留时间8.2min)。实验E8的色谱法的实验细节:制备分离在Agilent1100系列HPLC系统上进行,所述系统由chemstation/CC-模式软件包控制的Agilent1100脱气器、Agilent1100制备泵、Agilent1100二极管阵列检测器、Agilent1100MPSG2250A自动进样器/馏分收集器组成。制备分离的HPLC条件:柱:DaicelOD-H、250mm×20mm;洗脱剂0.5%异丙醇、正庚烷中的0.2%乙酸;流速13ml/min;检测220nm,400μl进样。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1