一种宝藿苷Ⅰ的制备方法与流程
本发明涉及宝藿苷Ⅰ的制备方法,属于食品、保健品、化妆品与医药领域。
背景技术:
宝藿苷Ⅰ,又名淫羊藿次苷Ⅱ(Icariside II),是从中药材淫羊藿中提取分离得到的主要活性成分淫羊藿苷经酶转化得到的新的有效单体,其结构式如下式(I)所示:
现有研究表明宝藿苷I具有改善心血管功能,调节内分泌和增强免疫力的作用。
该化合物的制备方法公开于200780034447.9,该方法以淫羊藿苷为原料,用可去除葡萄糖部分的酶或可以产生上述酶的微生物催化水解,反应产物经硅胶柱层析纯化,得到宝藿苷Ⅰ纯品。在硅胶柱层析纯化时,采用的溶剂分别为己烷和氯仿。
在申请号为201110186207.9的中国专利中公开了一种采用β-葡萄糖苷酶或纤维素酶酶解淫羊藿药材提取液,由于酶解得到的宝藿苷I含量很高,宝藿苷I难溶于水,因此需要在提取液中加入吐温,增加宝藿苷I在水中的溶解度,避免沉淀。后经大孔树脂色谱柱分离纯化,可获得纯度高于98%的宝藿苷Ⅰ。
在申请号为201210345102.8的中国专利中公开了一种宝藿苷Ⅰ的制备方法,该方法提供了一种从淫羊藿中同时提取分离淫羊藿苷和宝藿苷Ⅰ的方法,该方法将水提醇沉后的浓缩液,经过乙醇在大孔树脂梯度洗脱,再经过水-乙酸乙酯的萃取,收集乙酸乙酯层,再加入乙醇溶解过滤得到宝藿苷Ⅰ。
在申请号为200780034447.9中国专利所提供的方法中,宝藿苷Ⅰ的在纯化时,使用了毒性较大的氯仿进行提取;在申请号为201110186207.9的中国专利提供的方法中,需要采用吐温增加宝藿苷的溶解度,因此增加了制备成本;在申请号为201210345102.8的中国专利描述的方法为从淫羊藿中直接提取宝藿苷Ⅰ,由于宝藿苷I在淫羊藿药材中含量极低,一般不超过0.2%,因此,直接提取法将导致收率较低,对药材的浪费严重,不适于工业化生产。
技术实现要素:
本发明的一个目的是提供一种宝藿苷Ⅰ的制备方法,通过该方法制备得到的宝藿苷Ⅰ具有纯度高和收率高的优点。
本发明提供了一种宝藿苷Ⅰ的制备方法,该方法包括以下步骤:
a.将淫羊藿提取物在果胶酶的作用下进行酶解反应;
b.将酶解反应产物溶于弱极性有机溶剂,过滤,收集滤液;
c.在滤液中加入极性溶剂,结晶,得到宝藿苷I。
优选地,所述的淫羊藿提取物中淫羊藿苷的质量含量至少15%。
优选地,所述的弱极性有机溶剂选自乙酸乙酯、丙酮、二氯甲烷和甲基叔丁基醚中的一种或几种。
优选地,所述的弱极性有机溶剂为丙酮。
优选地,所述的极性溶剂为甲醇、乙醇和水中的一种或几种。
优选地,所述的极性溶剂为水。
优选地,在所述的步骤a和b之间还包括步骤d:将酶解反应得到的酶解产物离心,将离心得到的沉淀溶于弱极性有机溶剂。
优选地,在所述的步骤c中,在滤液中加入蒸馏水,进行回流,并将回流得到的液体冷却结晶。
优选地,滤液和蒸馏水的体积比为1-5:1,并且所述的回流温度为50-100℃,冷却结晶的温度为0-25℃。
更优选地,所述的滤液和蒸馏水的体积比为1-2:1,并且所述的回流 温度为50-80℃,冷却结晶的温度为10-20℃。
优选地,所述酶解反应的条件为:将淫羊藿提取物和磷酸缓冲液制成pH值为4.0-8.0的底物溶液,向底物溶液中加入果胶酶,果胶酶的质量为淫羊藿提取物质量的2-20倍,酶解温度为40-60℃,底物溶液中含有其总体积的0-25%的乙醇。
更优选地,果胶酶的质量为淫羊藿提取物质量的5-20倍,底物溶液的pH值为4.0-6.0,酶解温度为45-55℃。
最优选地,所述果胶酶的质量为淫羊藿提取物质量的15-20倍,底物溶液中含有其总体积的10-20%的乙醇。
优选地,所述底物溶液的pH值是通过磷酸缓冲液进行调节的。
更优选地,所述的磷酸缓冲液为磷酸-柠檬酸缓冲液或磷酸氢二钠-磷酸二氢钾缓冲液。
本发明的有益效果在于:本发明提供了一种与现有宝藿苷制备方法不同的宝藿苷的制备方法。通过本发明果胶酶水解淫羊藿提取物的收率高,相对于含有90%淫羊藿苷的淫羊藿提取物来说,宝藿苷Ⅰ的质量收率可以达到40%以上;通过果胶酶水解淫羊藿提取物的选择性好,产生的杂质少,只需要结晶工序便可得到纯度99%以上的宝霍苷I,易于工业化大生产。
附图说明
图1表示酶解反应进行12小时的高效液相色谱图。
图2表示酶解反应进行36小时的高效液相色谱图。
图3表示宝藿苷Ⅰ标准品的高效液相色谱图。
图4表示纯化后的宝藿苷Ⅰ的高效液相色谱图。
具体实施方式
除非另外说明,本文中的术语“酶解反应”指的是在酶作用下的水解反应。
除非另外说明,本文中术语的“淫羊藿提取物”指的是通过乙醇水溶剂提取淫羊藿得到的提取物,提取物中含有包括淫羊藿苷在内的黄酮类成分以及其他成分。
除非另外说明,本文中的术语“淫羊藿苷”指的是淫羊藿干燥茎叶中的提取物,为单一成分,具有如下分子结构:
除非另外说明,本文中的术语“果胶酶”购自帝斯曼公司的商标为RAPIDASE果胶酶作为发酵用酶,产品编号为984。
实施例1
1.酶解反应制备宝藿苷Ⅰ
取含淫羊藿苷90%的淫羊藿提取物10g,溶解于140mL pH=5.9,1/15M磷酸二氢钾-磷酸氢二钠缓冲液中,搅拌条件下加入60mL 95%乙醇,100mL果胶酶溶液,混合均匀后,待温度恒定在52~55℃开始计时,反应48小时后停止反应,反应液降温至30℃。
2.宝藿苷I的纯化
酶解反应结束后,将反应液进行离心,收集滤饼,干燥后重量为7.55g,粉碎后加入176g丙酮,搅拌1小时后过滤,向丙酮溶液中129g纯化水,加热回流至溶液澄清。然后将溶液缓慢降温至14℃后保温2小时,过滤,收集滤饼并干燥,得到宝霍苷I一次结晶产物6.04g。将宝霍苷I一次结晶产物粉碎后加入165g丙酮,搅拌1小时后过滤,向丙酮溶液中120g纯化水,加热回流至溶液澄清。溶液缓慢降温至14℃后保温2小时,过滤,收集滤饼并干燥,得到宝霍苷I二次结晶产物共计4.1g,相对于淫羊藿提取物的质量收率为54.3%。
实施例2
1.酶解反应制备宝藿苷Ⅰ
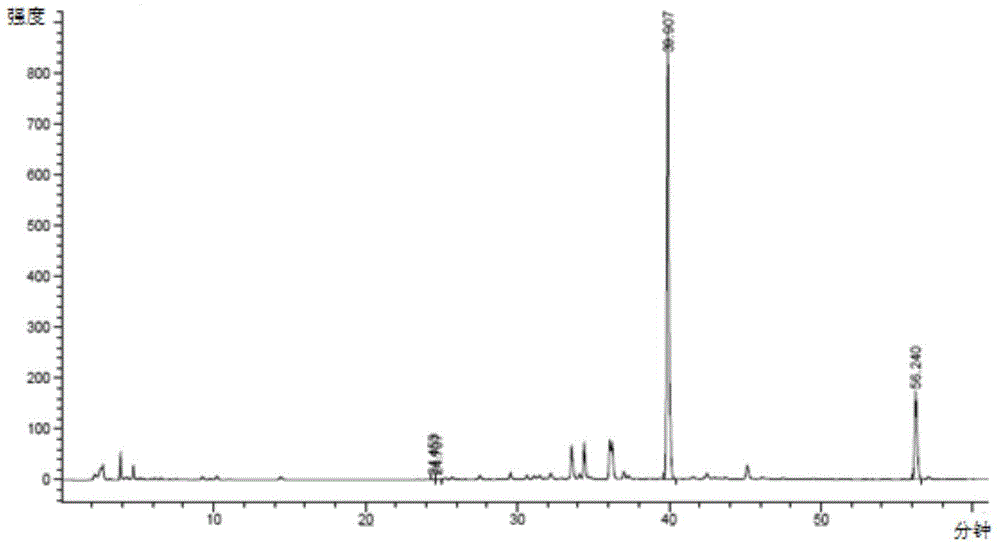
取含淫羊藿苷94%的淫羊藿提取物3.75kg,溶解于52L pH=5.9,1/15M磷酸二氢钾-磷酸氢二钠缓冲液中,搅拌条件下加入24L 95% 乙醇,75L果胶酶溶液,混合均匀后,待温度恒定在52~55℃开始计时,反应48小时后停止反应,反应液降温至30℃。反应12小时和36小时的高效液相色谱分别如图1和图2所示。通过图1和图2的比较,并对照图3,可以看出,随着酶解时间的增长,淫羊藿苷等底物相对于宝藿苷Ⅰ浓度下降,表明随着酶解时间的延长,淫羊藿苷等底物转化成宝藿苷I。
2.宝藿苷I的纯化
酶解反应结束后,反应液进行离心,收集滤饼,干燥后重量为2.8Kg,粉碎后加入66Kg丙酮,搅拌1小时后过滤,向丙酮溶液中48Kg纯化水,加热回流至溶液澄清。然后使溶液缓慢降温至14℃后保温2小时,过滤,收集滤饼并干燥,得到宝霍苷I一次结晶产物2.2Kg。将宝霍苷I一次结晶产物粉碎后加入60Kg丙酮,搅拌1小时后过滤,向丙酮溶液中44Kg纯化水,加热回流至溶液澄清。溶液缓慢降温至14℃后保温2小时,过滤,收集滤饼并干燥,得到宝霍苷I二次结晶产物共计1.5Kg,相对于淫羊藿提取物的质量收率为40%,纯度为99%,如图4所示。图1~4可以看出,酶解反应随着时间的延长,底物由淫羊藿苷逐步转化为宝藿苷I,反应生产含有宝藿苷I的酶解产物,经过丙酮-水两次结晶操作后,可将酶解产物中宝藿苷I的含量提高到99%,并且相对于酶解底物,宝藿苷I的产率可达到40%以上。
- 还没有人留言评论。精彩留言会获得点赞!