一种提高大肠杆菌异源合成聚酮类化合物的方法和用途与流程
本发明属于合成生物学和工业生物
技术领域:
,具体地说,本发明涉及一种提高大肠杆菌异源合成聚酮类化合物的方法和用途。
背景技术:
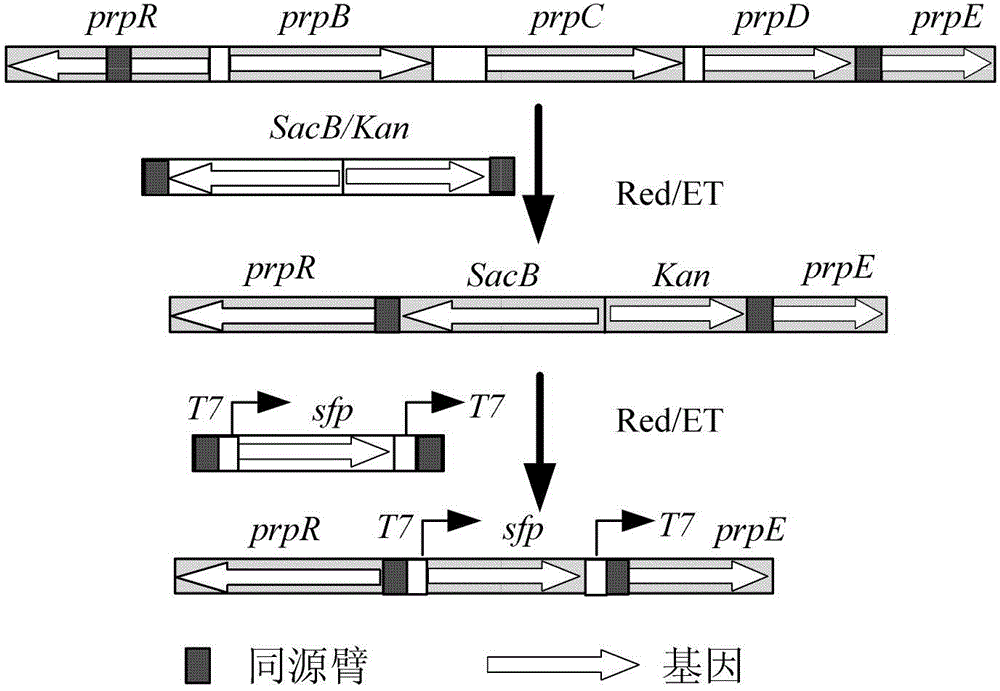
:天然产物在药物开发和发现过程中发挥重要作用。近十几年来,研究者使用异源合成天然产物表现出巨大的潜力。聚酮类化合物(Polyketids)的异源生物合成是目前合成生物学领域中研究较为深入、进展迅速的一个分支。Donadio于1991年提出了6-脱氧红霉素内酯B的合成模型,红霉素的聚酮合酶(PKS)研究因此而成为I型PKS的典范。红霉素生物合成分两个部分:第一是红霉素的母核6-脱氧红霉内酯B(6-deoxyerythronolide-B,6-dEB)的形成;第二是6-dEB侧链糖基化合成红霉素。6-dEB是红霉素合成过程中可分离出来的第一个中间体,合成6-dEB起始原料为丙酸和甲基丙二酸。整个过程由聚酮合酶催化完成。工程化的思想促使科研工作者构建一个工程菌进行异源合成天然产物。由于大肠杆菌代谢网络研究最为深入,因此大肠杆菌常作为工程菌的底盘细胞。2001年,Pfeifer等人利用大肠杆菌异源合成了红霉素第一个前体6-dEB,开启了异源生物合成红霉素的先河。以后的研究者通过大肠杆菌高效获得红霉素也做了诸多的尝试,如为了提高质粒稳定性,Murli等2003年用pccB和pccA基因整合到E.coli编码甲基丙二酰辅酶A脱羧酶ygfG位点,同时用RSF1010的复制起始位点替换pET28a来源于pBR322的起始位点,将pBP144改造为pKOS207-129,获得工程菌株K207-3/pKOS207-129/pBP130在摇瓶中可以获得22.5mg/L的6-dEB。为了提高宿主本身的稳定性,王勇等将红霉素聚酮合酶基因eryAI,AII,AIII通过染色体重组Red/ET方法整合入大肠杆菌染色体上,获得了染色体修饰的稳定菌株,与多个质粒共表达相比,该菌株可稳定的合成红霉素中间体6-dEB。尽管最近十几年中,以红霉素为代表的聚酮类化合物在大肠杆菌中的异源合成取得了一系列重大的进展,并在大肠杆菌中成功的合成了红霉素A。但仍存在着许多问题,如与原产株红霉糖多胞菌相比,红霉素在大肠杆菌中的产量过低。蒙海林等在2011年利用insilico分析研究平台对E.coli的代谢网络进行分析后发现目前大肠杆菌合成聚酮化合物的实际产量仅为理论产量的1/10左右,这说明异源的聚酮化合物合 成途径受大肠杆菌全局网络的调控。因此,对大肠杆菌进行全局网络改造,有可能进一步提高其异源合成聚酮化合物的能力。技术实现要素:本发明的目的在于提供一种提高大肠杆菌异源合成聚酮类化合物的方法和用途。在本发明的第一方面,提供一种促进合成聚酮类化合物6-脱氧红霉内酯的宿主菌生物合成聚酮类化合物6-脱氧红霉内酯的方法,所述方法包括:(1)在合成聚酮类化合物6-脱氧红霉内酯的宿主菌中,弱化靶基因表达;其中,所述的靶基因选自:(a)核苷酸合成和其它代谢模块基因purT、lsrC、hemN、zwf、pgl、gnd、rpe、talA、talB、tktA、tktB、ulaE或yieK;(b)磷酸戊糖和乙醛酸途径模块基因yaeR、rpiA、rpiB、purH、pyrB、pyrI、cysQ、pyrC、gmk、guaA、guaB、ndk、pyrF、pyrE、pyrH或hpt;(c)TCA循环和氧化磷酸化模块基因frdD、frdA、sdhA、sdhB、sdhC、sdhD、sucC、sucD、cyoA或cyoB;(d)糖代谢模块基因aceF、pgi、lpdA、ppk、ptsH、ptsI、glcF、glcE、fsaA或agaW;(e)6dEB前体代谢模块基因yjiM、scpA、scpB、tdcD、tdcE、pflB、pflD、PaaF、ackA、pta或ybiW;(f)脂肪酸代谢模块基因靶点fadJ、fadB、dhaK1、dhaK2或dhaH;(g)氨基酸和蛋白质合成代谢模块基因leuC、leuD、serC、serB、serA、gdhA或tnaA;或(h)frdD+sucC组合、lsrC+frdD组合、lsrC+sucC组合、frdD+rpiA组合、talA+guaB组合或zwf+guaB组合;(2)培养步骤(1)制备的菌株,从而生物合成聚酮类化合物6-脱氧红霉内酯。在一个优选例中,在合成聚酮类化合物6-脱氧红霉内酯的宿主菌中,弱化靶基因表达包括:引入抑制靶基因表达的干扰分子或敲除靶基因。在另一优选例中,所述的抑制靶基因表达的干扰分子针对(或自身具有):sucC中SEQIDNO:37所示的序列或其互补序列、tdcD中SEQIDNO:2所示的序列或其互补序列、scpB中SEQIDNO:3所示的序列或其互补序列、scpA中SEQIDNO:4所示的序列或其互补序列、ybiW中SEQIDNO:5所示的序列或其互补序列、pflB中SEQIDNO:6所示的序列或其互补序列、tdcE中SEQIDNO:7所示的序列或其互补序列、pflD中SEQIDNO:8所示的序列或其互补序列、paaF中SEQIDNO:9所示的序列或其互补序列、fadJ中SEQIDNO:10所示的序列或其互补序列、fadB中SEQIDNO:11所示的序列或其互补序列、ackA中SEQIDNO:12所示的序列或其互补序列、pta中SEQIDNO:13所示的序列或其互补序列、leuD中SEQIDNO:14所示的序列或其互补序列、leuC中SEQIDNO:15所示的序列或其互补序列、yjiM中SEQIDNO:16所示的序列或其互补序列、purT中SEQIDNO:17所示的序列或其互补序列、dhaK1中SEQIDNO:18所示的序列或其互补序列、dhaK2中SEQIDNO:19所示的序列或其互补序列、dhaH中SEQIDNO:20所示的序列或其互补序列、ptsH中SEQIDNO:21所示的序列或其互补序列、ptsI中SEQIDNO:22所示的序列或其互补序列、fsaA中SEQIDNO:23所示的序列或其互补序列、ppk中SEQIDNO:24所示的序列或其互补序列、aceF中SEQIDNO:25所示的序列或其互补序列、cyoA中SEQIDNO:26所示的序列或其互补序列、frdD中SEQIDNO:30所示的序列或其互补序列、frdA中SEQIDNO:31所示的序列或其互补序列、pgi中SEQIDNO:32所示的序列或其互补序列、sdhA中SEQIDNO:33所示的序列或其互补序列、sdhB中SEQIDNO:34所示的序列或其互补序列、sdhC中SEQIDNO:35所示的序列或其互补序列、sdhD中SEQIDNO:36所示的序列或其互补序列、sucD中SEQIDNO:38所示的序列或其互补序列、tnaA中SEQIDNO:39所示的序列或其互补序列、glcF中SEQIDNO:40所示的序列或其互补序列、glcE中SEQIDNO:41所示的序列或其互补序列、yaeR中SEQIDNO:42所示的序列或其互补序列、lsrC中SEQIDNO:43所示的序列或其互补序列、hemN中SEQIDNO:44所示的序列或其互补序列、agaW中SEQIDNO:45所示的序列或其互补序列、gdhA中SEQIDNO:46所示的序列或其互补序列、cyoB中SEQIDNO:47所示的序列或其互补序列、rpiA中SEQIDNO:48所示的序列或其互补序列、rpiB中SEQIDNO:49所示的序列或其互补序列、lpdA中SEQIDNO:50所示的序列或其互补序列、serC中SEQIDNO:51所示的序列或其互补序列、serB中SEQIDNO:28所示的序列或其互补序列、serA中SEQIDNO:29所示的序列或其互补序列、zwf中SEQIDNO:174所示的序列或其互补序列、pgl中SEQIDNO:175所示的序列或其互补序列、gnd中SEQIDNO:176所示的序列或其互补序列、rpe中SEQIDNO:177所示的序列或其互补序列、talA中SEQIDNO:178所示的序列或其互补序列、talB中SEQIDNO:179所示的序列或其互补序列、tktA中SEQIDNO:180所示的序列或其互补序列、tktB中SEQIDNO:181所示的序列或其互补序列、ulaE中SEQIDNO:182所示的序列或其互补序列、yieK中SEQIDNO:183所示的序列或其互补序列、purH中SEQIDNO:184所示的序列或其互补序列、pyrB中SEQIDNO:185所示的序列或其互补序列、pyrI中SEQIDNO:186所示的序列或其互补序列、cysQ中SEQIDNO:187所示的序列或其互补序列、pyrC中SEQIDNO:188所示的序列或其互补序列、gmk中SEQIDNO:189所示的序列或其互补序列、guaA中SEQIDNO:190所示的序列或其互补序列、guaB中SEQIDNO:191所示的序列或其互补序列、ndk中SEQIDNO:192所示的序列或其互补序列、pyrF中SEQIDNO:193所示的序列或其互补序列、pyre中SEQIDNO:194所示的序列或其互补序列、pyrH中SEQIDNO:195所示的序列或其互补序列、或hpt中SEQIDNO:196所示的序列或其互补序列。在另一优选例中,所述方法的(1)中,所述的靶基因选自:(a)核苷酸合成和其它代谢模块基因lsrC、zwf、pgl、gnd、rpe、talA、talB、tktA、tktB、ulaE或yieK;(b)磷酸戊糖和乙醛酸途径模块基因rpiA、purH、pyrB、pyrI、cysQ、pyrC、gmk、guaA、guaB、ndk、pyrF、pyrE、pyrH或hpt;(c)TCA循环和氧化磷酸化模块基因frdD、frdA、sdhA、sucC或sucD;(d)糖代谢模块基因ptsH、ptsI或glcE;(e)6dEB前体代谢模块基因yjiM、ackA、pta或ybiW;(f)脂肪酸代谢模块基因靶点fadB或dhaK2;(g)氨基酸和蛋白质合成代谢模块基因serC;或(h)frdD+sucC组合、lsrC+frdD组合、lsrC+sucC组合、talA+guaB组合或zwf+guaB组合。在另一优选例中,所述的抑制靶基因表达的干扰分子是sRNA。在另一优选例中,所述的sRNA包括以下结构:启动子、靶基因抑制分子(如靶基因结合序列)、终止子。在另一优选例中,所述的启动子选自:Pr启动子(较佳地,具有SEQIDNO:52中第7-61位的序列)、PBAD启动子、T7启动子、Trc启动子)。在另一优选例中,所述的终止子选自:TE终止子(较佳地,具有SEQIDNO:52中第171-310位的序列)、T1/TE终止子、T7终止子、rrnB终止子、rrnBT1和T2终止子)。在另一优选例中,所述抑制靶基因表达的干扰分子与终止子之间,还包括:micF序列(如具有SEQIDNO:52中第110-170位的序列)。在另一优选例中,所述的靶基因抑制分子是短核酸序列,例如长度为18-26bp;较佳地20-24bp;其能够与靶基因的mRNA互补或结合,或其具有靶基因mRNA的一段序列,在转入细胞内后可表达与靶基因的mRNA互补或结合的序列。在另一优选例中,所述的sRNA被包含在表达载体中。在另一优选例中,所述的合成聚酮类化合物6-脱氧红霉内酯的宿主菌是能合成聚酮类化合物6-脱氧红霉内酯的原核细菌。在另一优选例中,所述的能合成聚酮类化合物6-脱氧红霉内酯的原核细菌是能合成聚酮类化合物6-脱氧红霉内酯的大肠杆菌;较佳地,所述的大肠杆菌中,敲除了丙酸代谢操纵子并在该敲除位点上整合了磷酸泛酰巯基乙胺转移酶基因sfp;或直接敲除丙酸代谢操纵子,将sfp整合到大肠杆 菌基因组中任意一个非必须基因或无功能DNA序列区域。在另一优选例中,所述的大肠杆菌中转化有表达编码红霉素链霉菌聚酮合成酶DEBS2的基因、编码红霉素链霉菌聚酮合成酶DEBS3的基因、编码丙酰辅酶A羧化酶β-CT亚基的基因、编码丙酰辅酶A羧化酶a-CT亚基的基因和编码红霉素链霉菌聚酮合成酶DEBS1的基因。在本发明的另一方面,提供一种抑制靶基因表达的干扰分子,其是sRNA,所述的sRNA包括以下结构(较佳地,所述抑制靶基因表达的干扰分子与终止子之间,还包括:micF序列):启动子、靶基因抑制分子、终止子;在本发明的另一方面,提供所述的抑制靶基因表达的干扰分子如sRNA的用途,用于转化合成聚酮类化合物6-脱氧红霉内酯的宿主菌,弱化相应靶基因,促进合成聚酮类化合物6-脱氧红霉内酯的宿主菌生物合成聚酮类化合物6-脱氧红霉内酯。在本发明的另一方面,提供一种用于合成聚酮类化合物6-脱氧红霉内酯的宿主菌,所述的宿主菌中转化有抑制靶基因表达的干扰分子,或所述宿主菌中敲除了靶基因;其中,所述的靶基因选自:(a)核苷酸合成和其它代谢模块基因purT、lsrC、hemN、zwf、pgl、gnd、rpe、talA、talB、tktA、tktB、ulaE或yieK;(b)磷酸戊糖和乙醛酸途径模块基因yaeR、rpiA、rpiB、purH、pyrB、pyrI、cysQ、pyrC、gmk、guaA、guaB、ndk、pyrF、pyrE、pyrH或hpt;(c)TCA循环和氧化磷酸化模块基因frdD、frdA、sdhA、sdhB、sdhC、sdhD、sucC、sucD、cyoA或cyoB;(d)糖代谢模块基因aceF、pgi、lpdA、ppk、ptsH、ptsI、glcF、glcE、fsaA或agaW;(e)6dEB前体代谢模块基因yjiM、scpA、scpB、tdcD、tdcE、pflB、pflD、PaaF、ackA、pta或ybiW;(f)脂肪酸代谢模块基因靶点fadJ、fadB、dhaK1、dhaK2或dhaH;(g)氨基酸和蛋白质合成代谢模块基因leuC、leuD、serC、serB、serA、gdhA或tnaA;或(h)frdD+sucC组合、lsrC+frdD组合、lsrC+sucC组合、frdD+rpiA组合、talA+guaB组合或zwf+guaB组合。在另一优选例中,所述的宿主菌中转化有所述的抑制靶基因表达的干扰分子如sRNA;和/或所述的合成聚酮类化合物6-脱氧红霉内酯的宿主菌是能合成聚酮类化合 物6-脱氧红霉内酯的原核细菌;较佳地,所述的大肠杆菌中,敲除了丙酸代谢操纵子并在该敲除位点上整合了磷酸泛酰巯基乙胺转移酶基因sfp;较佳地,所述的大肠杆菌中转化有表达编码红霉素链霉菌聚酮合成酶DEBS2的基因、编码红霉素链霉菌聚酮合成酶DEBS3的基因、编码丙酰辅酶A羧化酶β-CT亚基的基因、编码丙酰辅酶A羧化酶a-CT亚基的基因和编码红霉素链霉菌聚酮合成酶DEBS1的基因。在本发明的另一方面,提供一种用于促进合成聚酮类化合物6-脱氧红霉内酯的宿主菌生物合成聚酮类化合物6-脱氧红霉内酯的试剂盒,所述的试剂盒中包括:所述的宿主菌的总和(本领域人员可以从中选择其中的一个或多个菌株用于生产);或所述的试剂盒中包括:所述的抑制靶基因表达的干扰分子的总和(本领域人员可以从中选择其中的一个或多个抑制靶基因表达的干扰分子进行应用);或所述的试剂盒中包括:分别包含所述的抑制靶基因表达的干扰分子的载体的总和(本领域人员可以从中选择其中的一个或多个载体进行应用)。本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。附图说明图1、sRNA表达质粒PJF650示意图。图2、E.coliWG构建过程示意图。图3、质粒pZG07构建过程。图4、质粒pZG08构建过程。图5、6-dEB分离组分的HPLC分析。图6、制备的6-dEB纯品NMR氢谱图。图7、40mg/L的6-dEBHPLC-ELSD分析图谱。图8、sRNA组合调控对大肠杆菌异源合成6-dEB影响。具体实施方式本发明人经过深入的研究,首次揭示一种促进合成聚酮类化合物6-dEB的宿主菌生物合成聚酮类化合物6-dEB的方法,通过单独弱化或组合弱化大肠杆菌中sucC(表达琥珀酸辅酶A合成酶)等基因的表达,来实现显著提高大肠杆菌异源合成聚酮类化合物的产量,最高产量提高率可达到60%以上。本发明的方法能实现发酵过程中聚酮类化合物大量积累。如本文所用,所述的“抑制靶基因表达的干扰分子”是指可以特异性降低靶基因表达水平的试剂,包括各种本领域已知可以抑制靶基因表达的分子,例如但不限于反义核酸,锁核酸,肽核酸,siRNA,shRNA,微小RNA等(统称为靶基因抑制分子);或携带或表达反义核酸,锁核酸,肽核酸,siRNA,shRNA,微小RNA等的构建物。如本文所用,所述的“异源”是指来自不同来源的两条或多条核酸或蛋白质序列之间的关系,获知来自不同来源的蛋白(或核酸)与宿主细胞之间的关系。例如,如果核酸与宿主细胞的组合通常不是天然存在的,则核酸对于该宿主细胞来说是异源的。特定序列对于其所插入的细胞或生物体来说是“异源的”。如本文所用,所述的“合成聚酮类化合物6-dEB的宿主菌”是指本领域已知可以应用于合成聚酮类化合物6-dEB的宿主菌或宿主细胞。所述的宿主菌包括但不限于:大肠杆菌,改进的大肠杆菌(包括转化入pccB和pccA的菌株,转化入eryAI,AII和/或AIII的菌株等)。优选的宿主菌是敲除了丙酸代谢操纵子并在该敲除位点上整合了磷酸泛酰巯基乙胺转移酶基因sfp的大肠杆菌,且其中转化有表达编码红霉素链霉菌聚酮合成酶DEBS2的基因、编码红霉素链霉菌聚酮合成酶DEBS3的基因、编码丙酰辅酶A羧化酶β-CT亚基的基因、编码丙酰辅酶A羧化酶a-CT亚基的基因和编码红霉素链霉菌聚酮合成酶DEBS1的基因。为了优化聚酮类化合物6-dEB的生成,本发明人作了广泛地研究,找到了适合于进行改良的靶基因,下调所述靶基因的表达有利于促进6-dEB的生成。本发揭示的各靶基因均是本领域已知的基因,本领域人员可以通过GenBank等平台查询到这些基因的序列信息,因此,本领域人员易于获得这些基因。本发明还提供了一个基因集合,所述基因集合包括:(a)核苷酸合成和其它代谢模块基因purT、lsrC、hemN、zwf、pgl、gnd、rpe、talA、talB、tktA、tktB、ulaE或yieK;(b)磷酸戊糖和乙醛酸途径模块基因yaeR、rpiA、rpiB、purH、pyrB、pyrI、cysQ、pyrC、gmk、guaA、guaB、ndk、pyrF、pyrE、pyrH或hpt;(c)TCA循环和氧化磷酸化模块基因frdD、frdA、sdhA、sdhB、sdhC、sdhD、sucC、sucD、cyoA或cyoB;(d)糖代谢模块基因aceF、pgi、lpdA、ppk、ptsH、ptsI、glcF、glcE、fsaA或agaW;(e)6dEB前体代谢模块基因yjiM、scpA、scpB、tdcD、tdcE、pflB、pflD、PaaF、ackA、pta或ybiW;(f)脂肪酸代谢模块基因靶点fadJ、fadB、dhaK1、dhaK2或dhaH;(g)氨基酸和蛋白质合成代谢模块基因leuC、leuD、serC、serB、serA、gdhA或tnaA。本领域技术人员根据本发明所揭示的内容,可以从该基因集合中选择一个或多个基因,进行如本发明所述的方法的操作,在所选的靶基因基础上设计下调其在宿主细胞中表达 的物质,从而用于促进6-dEB的生成。作为本发明的优选方式,所述的抑制靶基因干扰的干扰分子是sRNA或携带所述sRNA的构建物(含表达载体)。较佳地,所述的sRNA包括以下结构:启动子、靶基因抑制分子(如靶基因结合序列)和终止子;较佳地,所述靶基因抑制分子与终止子之间,还包括:micF序列。启动子和终止子的设计可以根据本领域技术人员的经验来进行,任何适用的启动子和终止子均包含在本发明的范围内。作为本发明的优选方式,所述的启动子是Pr启动子,所述的终止子是TE终止子。但是,本领域技术人员可以考虑对启动子和终止子作出变化,这仍然包含在本发明的范围内。通常,所述的sRNA位于表达载体上。因此,本发明还包括一种载体,它含有所述的sRNA。所述的表达载体通常还含有复制起点和/或标记基因等。本领域的技术人员熟知的方法能用于构建本发明所需的表达载体。这些方法包括体外重组DNA技术、DNA合成技术、体内重组技术等。应理解,只要能够实现sRNA的插入且转化细胞后能够下调靶基因的表达,任何表达载体均可选用的。此外,表达载体优选地包含一个或多个选择性标记基因,以提供用于选择转化的宿主细胞的表型性状。表达载体转化宿主细胞可用本领域技术人员熟知的常规技术进行。本发明还提供了用于合成聚酮类化合物6-dEB的宿主菌,所述的宿主菌中转化有抑制靶基因表达的干扰分子。所述的宿主菌可高效地生产聚酮类化合物6-dEB。本发明还提供了用于促进合成聚酮类化合物6-dEB的宿主菌生物合成聚酮类化合物6-dEB的试剂盒,所述的试剂盒中包括:转化有本发明的所有用于合成聚酮类化合物6-dEB的宿主菌的总和(本领域人员可以从中选择其中的一个或多个菌株用于生产);较佳地,所述的试剂盒中包括:转化有本发明更优选的用于合成聚酮类化合物6-dEB的宿主菌(转化有下调所述的靶基因或靶基因组合的基因表达的干扰分子的宿主菌)的总和。或者,所述的试剂盒中包括:本发明所述的sRNA的总和(本领域人员可以从中选择其中的一个或多个sRNA进行应用);较佳地,所述的试剂盒中包括:本发明更优选的sRNA(下调所述的靶基因或靶基因组合的基因表达的sRNA)的总和。所述的试剂盒中包括:包含有本发明所述的sRNA的载体的总和(本领域人员可以从中选择其中的一个或多个载体进行应用);较佳地,所述的试剂盒中包括:包含有本发明更优选的sRNA(下调所述的靶基因或靶基因组合的基因表达的sRNA)的载 体的总和。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如J.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。本发明的所使用的菌株、培养基及相关试剂如下:菌株E.coliDH10B作为克隆宿主,菌株E.coliWG(pZG07/pZG08)作为聚酮类化合物6-dEB合成的宿主(参见卢志国,华东理工大学博士学位论文,2011年)。分子克隆相关的酶、DNA片段与质粒抽提纯化试剂盒分别由NEB公司、TaKaRa公司及Axygen公司提供。各培养基组分、抗生素及其他相关试剂购自Oxiod、国药集团与上海生物工程有限公司。引物由南京金斯瑞生物科技公司合成。本发明中涉及的6-dEB发酵培养基配方(g/L):NaCl,10;蛋白胨,10;酵母提取物,5;甘油,15;100mMHEPES,pH调节为7.6。本发明中涉及的诱导剂:IPTG:24μg/ml;前体:丙酸钠20mM;本发明中涉及的诱导条件:22℃,250rpm,100ml摇瓶装液10ml培养基发酵培养5d。本发明中涉及的抗生素浓度分别为氨苄青霉素100mg/L,卡那霉素50mg/L,氯霉素34mg/L。实施例1、sRNA表达质粒的构建本发明是通过sRNA干扰技术进行调控靶点基因的表达,即表达sRNA与靶基因的mRNA结合,从而阻遏靶基因的mRNA与核糖体的结合,继而抑制靶基因的表达。本发明是通过对所选基因靶点进行弱化,鉴定出有利于提高合成聚酮类化合物的靶点基因。采用化学合成的deoB-sRNA基因序列片段(由上海捷端生物工程有限公司合成),包括Pr启动子、deoB靶基因结合位点(DNA序列长度为24bp,caccataataaatgcacgtttcat(SEQIDNO:1);与磷酸戊糖变位酶deoB基因从ATG起始序列开始后的24bp完全互补,deoB基因Genbank号:NC_012971.2)、micF序列(Genbank号:NC_000913.3)和TE终止子,并在两端引入NdeI和HindIII酶切位点。此sRNA基因序列片段DNA序列如下(SEQIDNO:52):catatgggatcctaacaccgtgcgtgttgactattttacctctggcggtgataatggttgccaccataataaatgcacgtttcattttctgttgggccattgcattgccactgattttccaacatataaaaagacaagcccgaacagtcgtccgggctttttttctcgagct cgagccaggcatcaaataaaacgaaaggctcagtcgaaagactgggcctttcgttttatctgtttttgtcggtgaacgctctctactagagtcacactggctcaccttcgggtgggcctttctgcgtttataactagtagatctaagcttsRNA表达质粒的构建以pACYCDuet-1质粒(购自Novagen)为PCR模板,通过引物pACYC-F和pACYC-R(引物两端引入NdeI和HindIII酶切位点)克隆得到只包含氯霉素抗性和p15A复制子的载体片段,应用清洁回收试剂盒(购自Axygen)回收后采用NdeI和HindIII双酶切此载体片段;同时采用NdeI和HindIII双酶切上述化学合成的deoB-sRNA序列片段。用清洁回收试剂盒直接回收双酶切后的载体片段和deoB-sRNA片段,然后通过T4连接酶将这两个回收片段连接,得到模板质粒pJF650(图1),用于弱化大肠杆菌中deoB基因的表达。以模板质粒pJF650为模板,采用表1中的引物和表2中的PCR条件,通过定点突变PCR扩增直接获得能够弱化大肠杆菌中(1)TCA循环和氧化磷酸化模块基因frdD、frdA、sdhA、sdhB、sdhC、sdhD、sucC、sucD、cyoA、cyoB;(2)糖代谢模块基因aceF、pgi、lpdA、ppk、ptsH、ptsI、glcF、glcE、fsaA、agaW;(3)6dEB前体代谢模块基因yjiM、scpA、scpB、tdcD、tdcE、pflB、pflD、PaaF、ackA、pta、ybiW;(4)磷酸戊糖和乙醛酸途径模块基因yaeR、rpiA、rpiB、purH、pyrB、pyrI、cysQ、pyrC、gmk、guaA、guaB、ndk、pyrF、pyrE、pyrH、hpt;(5)脂肪酸代谢模块基因靶点fadJ、fadB、dhaK1、dhaK2、dhaH;(6)氨基酸和蛋白质合成代谢模块基因leuC、leuD、serC、serB、serA、gdhA、tnaA;(7)核苷酸合成和其它代谢模块基因purT、lsrC、hemN、zwf、pgl、gnd、rpe、talA、talB、tktA、tktB、ulaE、yieK等不同靶点基因表达的sRNA质粒文库pSJ01(靶点基因tdcD)、pSJ02(靶点基因scpB)、pSJ03(靶点基因scpA)、pSJ04(靶点基因ybiW)、pSJ05(靶点基因pflB)、pSJ06(靶点基因tdcE)、pSJ07(靶点基因pflD)、pSJ08(靶点基因paaF)、pSJ09(靶点基因fadJ)、pSJ10(靶点基因fadB)、pSJ11(靶点基因ackA)、pSJ12(靶点基因pta)、pSJ13(靶点基因leuD)、pSJ14(靶点基因leuC)、pSJ15(靶点基因yjiM)、pSJ16(靶点基因purT)、pSJ17(靶点基因dhaK1)、pSJ18(靶点基因dhaK2)、pSJ19(靶点基因dhaH)、pSJ20(靶点基因ptsH)、pSJ21(靶点基因ptsI)、pSJ22(靶点基因fsaA)、pSJ24(靶点基因ppk)、pSJ26(靶点基因aceF)、pSJ29(靶点基因cyoA)、pSJ30(靶点基因frdD)、pSJ33(靶点基因frdA)、pSJ34(靶点基因pgi)、pSJ35(靶点基因sdhA)、pSJ36(靶点基因sdhB)、pSJ37(靶点基因sdhC)、pSJ38(靶点基因sdhD)、pSJ39(靶点基因sucC)、pSJ40(靶点基因sucD)、pSJ41(靶点基因tnaA)、pSJ43(靶点基因glcF)、pSJ44(靶点基因glcE)、pSJ50(靶点基因yaeR)、pSJ53(靶点基因lsrC)、pSJ54(靶点基因hemN)、pSJ66(靶点基因agaW)、pJF663(靶点基因zwf)、pJF666(靶点基因pgl)、pJF670(靶点基因gnd)、pSJ128(靶点基因rpe)、pSJ129(靶点基因talA)、pSJ130(靶点基因talB)、pSJ131(靶点基因tktA)、pSJ132(靶点基因tktB)、pSJ133(靶点基因ulaE)、 pSJ141(靶点基因yieK)、pSJ147(靶点基因purH)、pSJ148(靶点基因pyrB)、pSJ149(靶点基pyrI)、pSJ150(靶点基因cysQ)、pSJ151(靶点基因pyrC)、pSJ152(靶点基因gmk)、pSJ153(靶点基因guaA)、pSJ154(靶点基因guaB)、pSJ155(靶点基因ndk)、pSJ156(靶点基因pyrF)、pSJ157(靶点基因pyrE)、pSJ158(靶点基因pyrH)、pSJ159(靶点基因hpt)、pJF656(靶点基因gdhA)、pJF664(靶点基因cyoB)、pJF667(靶点基因rpiA)、pJF668(靶点基因rpiB)、pJF671(靶点基因lpdA)、pJF672(靶点基因serC)、pJF673(靶点基因serB)和pJF674(靶点基因serA)(表3)。以构建弱化sucC(表达琥珀酸辅酶A合成酶)靶基因的sRNA质粒为例,采用表1中的sucC-sRNA-F和sucC-sRNA-R引物及表2中的PCR条件,以模板质粒pJF650为模板,通过定点突变PCR直接将pJF650质粒骨架中24bp的deoB靶基因结合位点突变成sucC靶基因结合位点(与sucC基因从ATG起始序列开始后的24bpDNA序列完全互补),得到能够弱化sucC基因表达的sRNA质粒pSJ39。表1、质粒构建的引物表2、PCR条件表3、sRNA技术调控靶点功能实施例2、E.coliWG菌株的构建对E.coli敲除其丙酸代谢操纵子并在该位点上整合来自枯草芽孢杆菌(B.subtilis)表面素合成途径基因簇中的磷酸泛酰巯基乙胺转移酶基因sfp,构建适于聚酮化合物生物合成的E.coliWG菌株,其具体步骤如下:首先,以B.subtilis的基因组作为模板,采用引物sfp-F和sfp-R(表4)通过PCR扩增675bp的sfp基因(编码磷酸泛酰巯基乙胺转移酶,Genbank号:NC_000964),然后用NdeI和BamHI分别酶切PCR产物和pET21c质粒(购自Novagen公司)后,用T4DNA连接酶连接,构建质粒pET21c-sfp;然后,以质粒pUC19-sacB/kan作为模板,用引物SacB/Kan-F和SacB/Kan-R(表4)扩增带有同源臂的筛选标记基因的Kan-SacB片段(约2.8Kb),然后利用λRed/ET同源重组方法(Datsenkietal.PNAS,2000,97:6640-6645)将Kan-SacB整合到E.coliBL21(DE3)染色体的丙酸代谢操纵子中,替换丙酸代谢操纵子中prpR,prpB,prpC和prpD的DNA片段;利用WG-F和WG-R作为引物,扩增位于重组菌染色体上的SacB/Kan基因进行验证;然后,利用sfpR-F和sfpR-R从pET21c-sfp中扩增带有T7启动子的sfp基因;再次采用λRed/ET同源重组方法将重组菌中SacB-Kan片段替换为T7-sfp-T7片段,最终得到本发明所需的适用于聚酮化合物生物合成的E.coliWG菌株,其具体改构过程的示意图如图2所示。表4、本实例所用的引物及其所对应的碱基序列实施例3、质粒pZG07的构建以红霉糖多孢菌的基因组DNA为模板,利用引物eryAII-F、eryAII-R和eryAIII-F、eryAIII-R(表5)分别PCR扩增eryAII(编码红霉素链霉菌聚酮合成酶DEBS2,来源于Genbank号:NC_009142中)和eryAIII(编码红霉素链霉菌聚酮合成酶DEBS3,来源于Genbank号:NC_009142)基因,经纯化后的PCR产物分别用NdeI/HindIII双酶切,然后用T4DNA连接酶将双酶切的PCR产物分别与经同样酶切的质粒pET21c连接,分别获得质粒pZG05(含有eryAII)和pZG06(含有eryAIII);然后用XbaI/EcoRI酶切pZG06,回收含eryAIII基因的DNA片段后,用T4DNA连接酶将其与经SpeI/EcoRI酶切的质粒pZG05连接,构建质粒pZG07,所述的聚酮合成基因簇质粒pZG07的构建过程示意图如图3所示。表5、本实例所用的引物及其所对应的碱基序列实施例4、质粒pZG08的构建以天蓝色链霉菌的基因组DNA为模板,利用引物pccB-F,pccB-R和accA2-F,accA2-R(表6)分别扩增pccB基因(编码丙酰辅酶A羧化酶β-CT亚基,来源于Genbank号:NC_003888.3)和accA基因(编码丙酰辅酶A羧化酶a-CT亚基,来源于Genbank号:NC_003888.3),经纯化后的PCR产物分别用NcoI/EcoRI双酶切,然后用T4DNA连接酶将双酶切的PCR产物连接到经NcoI/EcoRI双酶切处理的pET28a质粒上,构 建得到质粒pZG01和pZG02;然后用XbaI/EcoRI双酶切处理pZG02,回收含基因accA2DNA片段,用T4DNA连接酶将其与经SpeI/EcoRI双酶切处理的pZG01质粒连接,得到质粒pZG03。以红霉糖多孢菌基因组DNA为模板,利用引物eryAI-F和eryAI-RPCR扩增eryAI基因(编码红霉素链霉菌聚酮合成酶DEBS1,Genbank号:NC_009142),经纯化后用NdeI/EcoRI双酶切处理,然后用T4DNA连接酶将其连接到经NdeI/EcoRI双酶切处理的质粒pET28a上,构建得到质粒pZG04;利用BglII对pZG04进行线性化并回收线性化片段,随后用核酸外切酶I对线性化片段末端进行消化,使BglII酶切后的粘性末端转变为平末端后再次回收该线性化片段,最后将该片段用HindIII消化后回收eryAI基因片段;对pZG03首先用EcoRI将其线性化并回收,随后用核酸外切酶I将EcoRI的粘性末端转变为平末端并回收,最后将线性化的pZG03用HindIII消化并回收。将上述回收后的pZG03和eryAI基因片段连接,构建质粒pZG08,所述的质粒pZG08的构建过程示意图如图4所示。表6、本实例所用的引物及其所对应的碱基序列实施例5、聚酮化合物6-dEB工程菌的构建及其5L罐发酵培养将质粒pZG07,pZG08共同转化到上述所得的聚酮化合物异源合成宿主菌E.coliWG后,得到能有效合成聚酮化合物的工程菌E.coliWG(pZG07/pZG08)(用于生产红霉素前体6-dEB)。挑取单菌落于2ml加有100mg/L羧苄青霉素和50mg/L卡那霉素的LB培养基中,220rpm,37℃培养过夜,得一级种子。然后将上述所得的一级种子以1%接种量接种到含有50ml的加有100mg/L羧苄青霉素和50mg/L卡那霉素的LB培养基的500ml摇瓶中,220rpm,37℃培养至OD600约为1,得二级种子。然后将所得的二级种子以1%接种量接种到含有4L的加有100mg/L羧苄青霉素和50mg/L卡那霉素的发酵培养基的5L罐中,同时加入终浓度为0.1mM的IPTG,20mM的丙酸钠于22℃,250rpm诱导培养5天,发酵结束。实施例6、6-dEB制备与HPLC-ELSD分析检测实施例5中发酵液用等体积的乙酸乙酯萃取3次,减压浓缩,得到粗浸膏。粗浸膏在反相C-18柱中利用甲醇水体系进行洗脱,甲醇比例分别是30%、50%、70%和100%。对分离组分利用Ultimate3000分析型HPLC分析。分析结果显示产物6-dEB主要存在于甲醇比例70%的组分中,HPLC分析见图5。利用反相C-18柱分离上述含6-dEB的粗组分,并用HPLC-ELSD分析制备,得到6-dEB纯品。HPLC-ELSD检测制备条件如下:70%甲醇洗脱下的组分,利用Ultimate3000制备型HPLC进行分离制备,色谱柱型号为:TSK-100V,5μm,19*150mm,流速15ml/min,流动相50%乙腈/水体系等度洗脱。ELSD检测器条件:蒸发光散射检测器漂移管温度:95℃;气体流速:1.6l/min;增益:16。保留时间tR:10.3min。对6-dEB纯品利用核磁分析验证,氢谱图见图6,从图可知,制备得到的纯品的H谱化学位移与文献报道一致(XinGao,SangKookWoo,MichaelJ.Krische,TotalSynthesisof6-DeoxyerythronolideBviaC-CBond-FormingTransferHydrogenation,J.Am.Chem.Soc.2013,135,4223-4226),确定此化合物为6-dEB。将制备的标准品配制成40mg/L的甲醇溶液,利用Ultimate3000分析型HPLC分析检测,条件:色谱柱TSK-100V,5μm,4.6*150mm;流速1ml/min;流动相乙腈/水,50%的乙腈梯度洗脱。ELSD为检测器,条件:蒸发光散射检测器漂移管温度:95℃;气体流速:1.6l/min;增益:16。从图7可知,制备得到6-dEB纯品有非常高的纯度,纯度达到98%以上。实施例7、sRNA质粒的转化与发酵将实施例1中的表达的各sRNA质粒(以pACYCDuet-1(购自Novagen公司)作为空白对照)分别转化到宿主细胞E.coliWG(pZG07/pZG08)(用于生产红霉素前体6-dEB)中,分别挑取单菌落至含羧苄青霉素(100mg/L),卡那霉素(50mg/L)和氯霉素(34mg/L)的2mlLB培养基中,37℃,250rpm/min过夜培养12h,作为摇床发酵培养种子。按1%接种量将上述种子接种于含10ml6-dEB发酵培养基(含羧卞青霉素,卡那霉素和氯霉素抗生素)的100ml摇瓶中,同时加入最终浓度为20mM的丙酸钠前体和100mM异丙基-β-D-硫代吡喃半乳糖苷(IPTG)初始诱导,每个样品平行三次。置于22℃,250rpm/min的摇床上发酵5d。发酵结束后,将发酵液倒入10ml离心管中,-20℃保存,用于后续的检测与分析。实施例8、实施例7中发酵液的6-dEB分析检测将实施例7中发酵液利用高效液相色谱-蒸发光散射检测器(HPLC-ELSD)进行分析检测。条件:色谱柱TSK-100V,5μm,4.6*150mm;流速1ml/min;流动相乙腈/水,50%的乙腈等度洗脱。ELSD为检测器,条件:蒸发光散射检测器漂移管温度:95℃;气体流速:1.6l/min;增益:16。不同基因弱化对6-dEB合成的影响结果见表7(以对照组E.coliWG(pZG07/pZG08/pACYCDunet-1)的6-dEB产量为100%)。由表7可知:利用sRNA对这些关键靶点基因弱化都是有利于合成目标产物6-dEB。例如,菌株E.coliWG(pZG07/pZG08/pSJ39),其调控的关键靶点基因为sucC(合成酶:琥珀酰辅酶A合成酶的β亚基,succinyl-CoAsynthetaseβsubunit)。相对于对照组E.coliWG(pZG07/pZG08/pACYCDunet-1),弱化sucC可使宿主合成6-dEB产量提高率达到63.2%。提高产量最高的是E.coliWG(pZG07/pZG08/pSJ130),其调控的关键靶点基因为talB(转醛醇酶,transaldolase),弱化talB可使宿主合成6-dEB产量提高率达到1008.81%。采用sRNA方法调控大肠杆菌底盘细胞的代谢网络,通过弱化这些基因能显著提高聚酮化合物的异源合成。通过弱化提高聚酮化合物产量超过20%以上的靶点有:ybiW、fadB、ackA、pta、yjiM、dhaK2、ptsH、ptsI、frdD、frdA、sdhA、sucC、sucD、glcE、lsrC、rpiA、serC、talA、talB、zwf、pyrI、cysQ、gmk、guaB、pyrH、hpt。表7、sRNA干扰对异源宿主E.coli合成6-dEB的影响实施例9、sRNA质粒共转组合弱化进一步提高聚酮类化合物6-dEB产量通过实施例8中的数据可知:选取弱化后提高聚酮化合物产量超过20%以上的靶点ybiW、fadB、ackA、pta、yjiM、dhaK2、ptsH、ptsI、frdD、frdA、sdhA、sucC、sucD、glcE、lsrC、rpiA、serC,将以上这些弱化质粒的氯霉素抗性替换为阿普拉霉素抗性,用于后续组合共转的抗性筛选。替换抗性质粒构建是基于POE-PCR技术方法(Youetal.SimplecloningviadirecttransformationofPCRproduct(DNAMultimer)toEscherichiacoliandBacillussubtilisApplEnvironMicrobiol,78(5):1593-1595),以替换对照质粒pACYCDuent-1的抗性为例,首先,采用表8中Aparamycin-F和Aparamycin-R引物及PCR条件(见表9),以pKC1139(链霉菌通用质粒)为模板,通过PCR直接将pKC1139质粒中阿普拉霉素抗性基因扩增出来;采用表8中sRNA-Aparamycin-F和sRNA-Aparamycin-R引物及PCR条件,以pACYCDuent-1为模板,通过PCR将pACYCDuent-1质粒除氯霉素抗性基因以外的序列扩增出来。接着采用表9中PCR条件(注:不用加引物,模板为第一步扩增出来的阿普拉霉素抗性基因和不含氯霉素抗性基因的pACYCDuent-1,各1μl),进行PCR,将此PCR产物用PCR清洁回收试剂盒回收后,直接转化到DH10B中,涂布于含50mg/L阿普拉霉素的LB固体培养基平板上过夜培养。挑取单菌落于2ml含50mg/L阿普拉霉素的LB培养基中,220rpm,37℃培养过夜,采用质粒提取试剂盒提取质粒,得到质粒pSJ77。表8、引物信息和PCR条件表9、PCR条件其余的表达sRNA质粒替换抗性同上,采用相同引物和PCR条件将质粒上的氯霉素抗性替换为阿普拉霉素抗性,各sRNA质粒信息如表10。表10、sRNA质粒信息将上述构建表达sRNA质粒(以pSJ77作为空白对照)和相对产量提高20%的实施例1构建的表达sRNA质粒两两组合,共转到宿主细胞E.coliWG(pZG07/pZG08)(用于生产红霉素前体6-dEB),利用实施例7的发酵方法和实施例8中的6-dEB分析检测。sRNA组合发酵实验以对照组E.coliWG(pZG07/pZG08/pSJ39/pSJ77)的6-dEB产量为100%,通过组合弱化关键靶点基因来进一步提高聚酮类化合物6-dEB的产量。组合sRNA干扰对异源宿主E.coli合成6-dEB的影响的实验结果如表11。表11、组合sRNA干扰对异源宿主E.coli合成6-dEB的影响实验结果表明,尽管多数组合弱化不能有效提高聚酮类化合物的合成,但组合弱化frdD和sucC可使6-dEB产量得到进一步提高,比单独弱化sucC的产量再提高24%以上,同时弱化lsrC和frdD可使6-dEB产量进一步提高17%以上。实施例10、弱化两靶点的sRNA质粒进一步提高聚酮类化合物6-dEB产量根据实施例8中的数据可知:提高聚酮化合物产量超过550%的弱化靶点有:核苷酸合成和其它代谢模块基因talA、talB、zwf和磷酸戊糖和乙醛酸途径模块基因pyrI、cysQ、gmk、guaB、pyrH、hpt,考察同时弱化这两个模块的靶点对合成聚酮类化合物6-dEB产量的影响。弱化两靶点的sRNA质粒均采用酶切串联的方法进行构建。以构建pSJ333为例:以pSJ130为模板,以sRNA-F/R为引物(表12)扩增靶向talB的sRNA骨架,PCR条件以实施例9中表9相同。清洁回收PCR产物后,用BamHI和HindIII酶切;同时用BglII和HindIII酶切pSJ129为载体,清洁回收。将上述PCR酶切产物和pSJ129 双酶切产物用T4连接酶连接,得到质粒pSJ333。其余质粒利用相同的方式获得,最终得到质粒pSJ404-pSJ421,质粒信息见表13。表12引物信息Primer名称序列(5’→3’)SRNA-FCGGGATCCTAACACCGTGCGTGTTGACTATTTTASRNA-RCCCAAGCTTAGATCTACTAGTTATAAACGCAGAAAGG表13组合sRNA质粒信息名称sRNA技术调控(弱化)靶点抗性pSJ333talA+talB氯霉素pSJ334cysQ+guaB氯霉素pSJ404talA+pyrI氯霉素pSJ405talA+cysQ氯霉素pSJ406talA+gmk氯霉素pSJ407talA+guaB氯霉素pSJ408talA+pyrH氯霉素pSJ409talA+hpt氯霉素pSJ410talB+pyrI氯霉素pSJ411talB+cysQ氯霉素pSJ412talB+gmk氯霉素pSJ413talB+guaB氯霉素pSJ414talB+pyrH氯霉素pSJ415talB+hpt氯霉素pSJ416zwf+pyrI氯霉素pSJ417zwf+cysQ氯霉素pSJ418zwf+gmk氯霉素pSJ419zwf+guaB氯霉素pSJ420zwf+pyrH氯霉素pSJ421zwf+hpt氯霉素将上述构建表达组合sRNA质粒(以pACYCDuent-1作为空白对照)转到宿主细胞 E.coliWG(pZG07/pZG08)(用于生产红霉素前体6-dEB),利用实施例7的发酵方法和实施例8中的6-dEB分析检测。sRNA组合发酵实验以对照组E.coliWG(pZG07/pZG08/pSJ39/pSJ77)的6-dEB产量为100%,通过组合弱化关键靶点基因来进一步提高聚酮类化合物6-dEB的产量。组合sRNA干扰对异源宿主E.coli合成6-dEB的影响的实验结果如表14和图8。实验表明,尽管所有组合弱化都能有效提高聚酮类化合物的合成,但只有组合弱化talA+guaB和zwf+guaB可使6-dEB产量得到进一步提高,比单独弱化talB的产量再提高32%以上,比对照组的产量再提高1300%以上,使6-dEB摇瓶产量可达到210mg/L以上。表14、组合sRNA干扰对异源宿主E.coli合成6-dEB的影响质粒名称sRNA质粒弱化的靶点6-dEB产量(mg/L)6-dEB相对产量提高率(%)pACYCDuent-1control14.300.00pSJ404talA+pyrI135.10844.76pSJ405talA+cysQ128.87801.19pSJ406talA+gmk117.09718.81pSJ407talA+guaB209.381364.20pSJ408talA+pyrH153.30972.03pSJ409talA+hpt118.84731.05pSJ410talB+pyrI145.38916.64pSJ411talB+cysQ120.11739.93pSJ412talB+gmk138.00865.03pSJ413talB+guaB60.05319.93pSJ414talB+pyrH149.79947.48pSJ415talB+hpt100.32601.54pSJ416zwf+pyrI152.75968.18pSJ417zwf+cysQ130.45812.24pSJ418zwf+gmk119.39734.90pSJ419zwf+guaB210.421371.47pSJ420zwf+pyrH75.35426.92在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单 独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。序列表当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1