氘代脱氢苯基阿夕斯丁类化合物及其制备方法和在制备抗肿瘤的药物中的应用与流程
本发明属于化学和医药技术领域,涉及医药化合物及其制备方法和应用,具体涉及氘代脱氢苯基阿夕斯丁类化合物及其制备方法和在制备抗肿瘤的药物中的应用。
背景技术:
众所周知,血管生成对肿瘤的生长和转移至关重要,因此肿瘤血管系统已成为极具价值的肿瘤治疗靶。一般说来,靶向肿瘤脉管系统的抗肿瘤药物主要包括抗血管生成剂和血管阻断剂(VDAs),前者可抑制肿瘤新血管的生长,而后者则是靶向破坏为肿瘤细胞供应氧和营养的已有血管网。肿瘤血管具有与正常血管不同的异常结构,其内皮细胞增殖迅速,血管壁变薄而具高渗透性,导致高血流阻力。VDAs则可选择性作用于肿瘤血管内皮细胞,干扰其黏连结合部位和微管骨架,从而影响内皮细胞的迁移、附着和增殖,导致血管的蛋白渗透性和间隙压力增加,引起血浆渗漏、血管直径减小和血液黏滞度提高,最终致使血流进一步减慢,氧和营养供应阻断,导致肿瘤坏死。
VDAs可分为配体靶向型VDAs和小分子VDAs两种类型,前者由靶向部分(如抗体、肽或生长因子)和效应子部分(可通过不同机制杀死肿瘤)组 成,后者又可进一步分为黄酮类和微管蛋白结合剂。由美国Nereus制药公司研制的Plinabulin(KPU-2,NPI-2358,中文名称:普那布林)便是一种微管蛋白结合剂,是源自海洋曲霉菌的低分子环二肽Phenylahistin或halimide的合成衍生物,可选择性作用于内皮微管蛋白中秋水仙碱结合位点附近,抑制微管蛋白聚合,阻断微管装配,从而破坏内皮细胞骨架,抑制肿瘤血流,但不伤害正常血管系统。
Plinabulin结合到微管蛋白的秋水仙素结合位点附近,作用于MM细胞,使MM细胞停在有丝分裂早期,而诱导细胞死亡。Plinabulin也抑制微管形成,及内皮细胞和MM细胞的迁移,使肿瘤脉管系统功能失常。Plinabulin可诱导癌细胞死亡,但不影响其他正常单核细胞的活力。
Plinabulin的化学结构式如下:
Plinabulin分子式为C19H20N4O2,分子量336.39,CAS号714272-27-2。具有较好的稳定性。
氘是氢的一种稳定非放射性同位素,重量为2.0144。由于生成的氘代化合物中的氘含量远远高于自然界中0.015%的含量,所以可以将其看做是一种新型的化合物。氘代对于药物的改善也得到了美国专利与商标局的认可,如氘代利莫那班(Rimonabant)、莫沙比利(Mosapride)、奥昔布宁(Oxybutynin)等专利都已经被批准。
研究表明,氘在药物设计中是目前最好的生物电子等排体,并且氘代 作用可以减少种群和性别不同引起的药效和毒性差异、减少病人间的个体差异、通过减少有害代谢物降低不良反应事件的发生、还可以减少基因毒性等,因此氘代作用已被广泛应用于人类临床研究以及药物研发过程中的药代动力学研究。目前国际上已有多家制药企业在从事氘代新药的研发,并已有多个氘代药物进入临床实验,如CTP-347、CTP-499等。
氘的重要特点是其在药物分子中的形状和体积与氢基本上相同,如果药物分子中的氢被选择性的替换为氘,氘代药物一般还会保留原来的生物活性和选择性。实验证明,碳-氘键的结合比碳-氢键更加稳定。携带中子的氘与碳形成的碳-氘键在较低的频率振动,因而强于碳-氢键。这一强度的增加,可直接影响某些药物的吸收、分布、代谢和排泄等属性,从而提高药物的疗效、安全性和耐受性。因此理论认为,如果药物分子中将被分解的某个特定的碳-氢键被氘代为相应的碳-氘键后,将会延缓其分解过程,使氘代药物在身体里作用的时间更长,成为效果更优于原来的药物。
技术实现要素:
本发明的目的在于提供了氘代脱氢苯基阿夕斯丁类化合物及其制备方法和在制备抗肿瘤的药物中的应用,本发明所提供的氘代脱氢苯基阿夕斯丁类化合物具有抑制肿瘤细胞的作用,可以用于制备抗肿瘤的药物。
为实现上述发明目的,本发明采用以下技术方案予以实现:
氘代脱氢苯基阿夕斯丁类化合物,其具有通式(I)所示结构:
其中R1为苯环上的一取代或多取代的取代基,所述R1选自氢、氘、卤素、羟基、甲氧基、氨基、苯基、氨基甲基苯基、C1-C24烷基、C2-C24烯基、C2-C24炔基、芳基烷基、杂环芳基烷基、C1-C24的酰基、C1-C24的烷氧基、羧基、羧酸酯基、酰胺基、N-单取代或N,N-双取代酰胺基、磺酸基、磺酸酯基、磺酰胺基、N-取代磺酰胺基、烷氧基、芳基烷氧基、烷硫基、氰基、氨基、取代的氨基、硝基、环烷基、环烯基、芳香基、取代的芳香基、芳香杂环基、芳氧基、芳酰基、环氧基、环酰基、芳香硫基或芳磺酰基中的至少一种;
R2为氢或氘,R3为氢或氘;
X1为氧或硫;
X2为NH、氧或硫。
进一步的:所述氘代脱氢苯基阿夕斯丁类化合物具体为:
(3Z,6Z)-3-苯基氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯基)氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-苯基氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代 亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-苯亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(4-氟-2,3,5,6-四氘代苯基)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(4-氟-2,3,5,6-四氘代苯基)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(3-氟代苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(3-苯甲酰基苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(3-(4-氟苯甲酰基)苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(3-(4-甲氧基苯甲酰基)苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(3Z,6Z)-3-(3-甲氧基苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
或(3Z,6Z)-3-(3-三氟甲基苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮。
本发明还提供了合成所述氘代脱氢苯基阿夕斯丁类化合物的氘醛中 间体,其具有通式(Ⅱ)所示结构:
其中X为NH、氧或硫。
进一步的:所述氘醛中间体具体为:5-叔丁基-1H-咪唑-4-氘代甲醛;5-叔丁基-噁唑-4-氘代甲醛或5-叔丁基-噻唑-4-氘代甲醛。
本发明还提供了合成所述氘代脱氢苯基阿夕斯丁类化合物的含氘杂环化合物中间体,其具有通式(Ш)所示结构:
X1为氧或者硫,X2为NH、氧或者硫;
其中取代基R选自C1-C24烷基、C2-C24烯基、C2-C24炔基、芳基烷基、杂环芳基烷基、C1-C24的酰基、磺酰基、芳酰基、环酰基、芳磺酰基中的其中至少一种。
进一步的:所述含氘杂环化合物中间体具体为:
(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(Z)-1-乙酰基-3-((5-叔丁基-噁唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(Z)-1-乙酰基-3-((5-叔丁基-噻唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(Z)-1-苯甲酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(Z)-1-烯丙基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
(Z)-1-甲磺酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮;
或(Z)-1-对甲苯磺酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮。
本发明提供了氘代脱氢苯基阿夕斯丁类化合物的制备方法,它包括以下步骤:
(1)将醛中间体a进行还原和氧化反应得到氘醛化合物b,所述醛中间体a和氘醛化合物b结构式如下:
(2)第一步缩合反应:先将二乙酰基哌嗪二酮与所述醛中间体a或者氘醛化合物b发生缩合反应,形成杂环化合物c或者含氘杂环化合物d;
所述杂环化合物c或者含氘杂环化合物d结构式如下:
(3)第二步缩合反应:再将所述杂环化合物c或者含氘杂环化合物d与第二缩合反应醛发生缩合反应形成氘代脱氢苯基阿夕斯丁类化合物;所述第二缩合反应醛为苯甲醛、氘代苯甲醛或苯环上带有取代基的苯甲醛衍生物、氘代苯甲醛衍生物。
进一步的:所述醛中间体a为咪唑甲醛,所述氘醛化合物b为氘代咪唑甲醛。
进一步的:所述步骤(1)中氘醛化合物b的制备方法为:将5-叔丁基-1H-咪唑-4-甲醛经由NaBD4还原和二氧化锰氧化,得到5-叔丁基-1H-咪唑-4-氘代甲醛为氘醛化合物b。
进一步的:所述步骤(2)中含氘杂环化合物d的制备方法为:将二乙酰基哌嗪二酮与所述氘醛化合物b在DMF作溶剂、碳酸铯作碱、N,N-二甲基甲酰胺作溶剂的条件下避光发生缩合反应,形成含氘杂环化合物d。
药物组合物,含有所述的化合物和药物可接受的载体。
本发明最后提供了所述的氘代脱氢苯基阿夕斯丁类化合物在制备抗肿瘤的制剂中的应用。
所述肿瘤包括皮肤癌和白血病。
本发明的优点和技术效果是:本发明提供了氘代脱氢苯基阿夕斯丁类化合物,该类化合物合成方法包括:先将二乙酰基哌嗪二酮与醛中间体a或者氘醛化合物b发生缩合反应,形成杂环化合物c或者含氘杂环化合物d,再与苯甲醛、氘代苯甲醛类化合物发生缩合反应形成氘代脱氢苯基阿夕斯丁类化合物。同时,本发明也提供了一种高效的高氘代率的氘醛中间体及含氘杂环中间体及其合成方法。经实验证明本发明提供的氘代脱氢苯基阿夕斯丁类化合物具有影响微管蛋白解聚,治疗难治性实体瘤或淋巴瘤的作用,本发明提供了研究与开发所述相关化合物的抗肿瘤药物的方法。
附图说明
图1表明了本发明所述化合物1-9在血浆中的代谢稳定性;
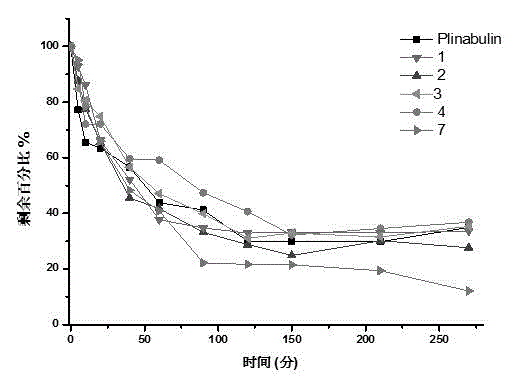
图2表明了本发明所述化合物1、2、3、4、7的体外肝微粒体代谢稳定性;
图3表明了Plinabulin(脱氢苯基阿夕斯丁)及其氘代类似物化合物1、2、3对A431细胞微管蛋白的影响;
图4表明了Plinabulin(脱氢苯基阿夕斯丁)及其氘代类似物化合物1、2、3对A431细胞微管蛋白荧光强度影响。
具体实施方式
以下结合具体实施例对本发明的技术方案做进一步详细的说明。
已知化合物5-叔丁基-1H-咪唑-4-甲醛(醛中间体a)的合成:
其合成步骤为,第一步是关环成噁唑环;第二步是在甲酰胺中加热处理,成为咪唑环;再经过还原并氧化得到咪唑醛类化合物,反应式如下:
其具体制备过程包括以下步骤:
(1)将异氰基乙酸乙酯(5.8mL,53mmol)溶于90mL干燥的四氢呋喃中,加入DBU(9.5mL,63.6mmol),然后慢慢加入三甲基乙酸酐(12.9mL,63.6mmol),室温下反应过夜,旋干溶剂,乙酸乙酯萃取,10%碳 酸钠溶液淋洗,再用10%柠檬酸溶液淋洗,无水硫酸钠干燥。旋干溶剂,柱层析分离纯化,石油醚:乙酸乙酯=15:1为洗脱剂,得到噁唑化合物10.3g,产率99%。
(2)在250mL圆底瓶中加入噁唑化合物(8.1g,41mmol),随后加入60mL甲酰胺,165℃下加热反应,24h后加入10%碳酸钠淬灭反应,乙酸乙酯萃取,无水硫酸钠干燥。旋干溶剂,柱层析分离纯化,二氯甲烷:甲醇=60:1为洗脱剂,得到咪唑化合物4.4g,产率55%。
(3)将咪唑化合物(830mg,4.2mmol)溶于干燥的四氢呋喃中,-5℃下加入四氢铝锂(479mg,12.6mmol),0.5h后升到室温反应4h,加水淬灭,砂芯漏斗抽滤,滤液旋干直接进行下一步反应。把旋干后的还原产物溶于20mL干燥的丙酮中,加入二氧化锰(3.6g,42mmol),室温下反应过夜,砂芯漏斗抽滤,旋干溶剂得到化合物5-叔丁基-1H-咪唑-4-甲醛351mg,两步产率55%。
已知化合物杂环中间体Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(杂环化合物c)的合成:
其制备方法为:将5-叔丁基-1H-咪唑-4-甲醛(304mg,2mmol),1,4-二乙酰基哌嗪-2,5-二酮(792mg,4mmol),DMF(9mL),碳酸铯(977mg,3mmol)置于反应瓶中,氮气保护,室温下反应18小时(TLC检测反应完全)。将反应液倒入冷水中,有固体析出,过滤,得浅黄色固体320mg, 即为所述(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮,收率为55%。
本发明所述氘代脱氢苯基阿夕斯丁类化合物的制备方法包括以下步骤:
(1)氘醛化合物b的合成:从醛中间体a出发,经由NaBD4还原和二氧化锰氧化,得到氘醛化合物b,
(2)第一步缩合反应:先将二乙酰基哌嗪二酮(DKP)与所述醛中间体a或者氘醛化合物b发生缩合反应,形成杂环化合物c或者含氘杂环化合物d;
(3)第二步缩合反应:再将所述杂环化合物c或者含氘杂环化合物d与第二醛,在避光条件下发生缩合反应,形成氘代脱氢苯基阿夕斯丁类化合物;所述第二醛为苯甲醛、氘代苯甲醛或苯环上带有取代基的苯甲醛衍生物、氘代苯甲醛衍生物。
实施例1
中间体5-叔丁基-1H-咪唑-4-氘代甲醛(氘醛化合物b)的制备,结构式如下:
其具体制备过程包括以下步骤:称取5-叔丁基-1H-咪唑-4-甲醛(304mg,2mmol)于50mL干燥单口瓶中,置换氮气保护。向反应瓶中加入干燥乙醇5mL,硼氘化钠(420mg,10mmol),置换氮气保护,室温下反应过夜,加水10mL淬灭反应,乙酸乙酯30mL萃取有机相,旋干后直接投入下步反应,加入10mL干燥的丙酮和二氧化锰(1.7g,20mmol),室温下反应过夜,砂芯漏斗抽滤,滤液旋干柱层析纯化,得到5-叔丁基-1H-咪唑-4-氘代甲醛(199mg,1.3mmol),为白色固体,产率65%。
1H NMR(400MHz,DMSO-d6)δ6.97(s,1H),6.66(s,1H)1.05(s,9H);MS(ESI)m/z 154.10(M+H)+(calcd forC8H12DN2O154.10)。
实施例2
(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮(含氘杂环化合物d)的制备,结构式如下:
其制备方法为:将5-叔丁基-1H-咪唑-4-氘代甲醛(306mg,2mmol),1,4-二乙酰基哌嗪-2,5-二酮(792mg,4mmol),无水硫酸镁(800mg), DMF(9mL),碳酸铯(977mg,3mmol)置于反应瓶中,氮气保护,室温下反应18小时(TLC检测反应完全)。将反应液倒入冷水中,有固体析出,过滤,得灰白色固体306mg,即为所述(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮,收率为52.58%。
1H NMR(400MHz,DMSO-d6)δ12.37(s,1H),12.00(s,1H),7.85(s,1H),4.31(s,2H),2.48(s,3H),1.39(s,9H);MS(ESI)m/z 292.14(M+H)+(calcd forC14H18DN4O3292.14)。
实施例3
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮的制备(化合物1)
所述化合物1的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮(250mg,0.86mmol),2,3,4,5,6-五氘代苯甲醛(191mg,1.72mmol),无水硫酸镁(350mg),DMF(8mL),碳酸铯(420mg,1.29mmol)置于反应瓶中,置换氮气保护,45℃反应24小时,TLC检测反应完全。将反应液加入到冷水中,有黄色固体析出,过滤,滤饼用无水乙醇和乙酸乙酯的混合溶剂溶解,滤去不溶物,减压浓缩至干,黄色固体140mg即为所述化合物1,收率47.54%。
1H NMR(400MHz,DMSO-d6)δ12.31(s,1H),12.22(s,1H),9.98(s, 1H),7.83(s,1H),6.74(s,1H),1.37(s,9H);MS(ESI)m/z 343.2045(M+H)+(calcd forC19H15D6N4O2343.2036)。
实施例4
(3Z,6Z)-3-苯基氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮的制备(化合物2)
所述化合物2的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮(200mg,0.69mmol),苯氘代甲醛(147mg,1.38mmol),无水硫酸镁(280mg),DMF(5mL),碳酸铯(336mg,1.03mmol)置于反应瓶中,氮气保护,45℃反应22小时,TLC检测反应完全。将反应液加入到冷水中,有黄色固体析出,过滤,滤饼用无水乙醇和乙酸乙酯的混合溶剂溶解,过滤,减压浓缩至干,得黄色固体75mg即为所述化合物2,收率32.26%。
1H NMR(400MHz,DMSO-d6)δ12.31(s,1H),12.22(s,1H),10.00(s,1H),7.83(s,1H),7.52(d,J=4Hz,2H),7.39(t,J=8Hz,2H),7.32(t,J=8Hz,1H),1.38(s,9H);MS(ESI)m/z 339.1788(M+H)+(calcd forC19H19D2N4O2339.1785)。
实施例5
(3Z,6Z)-3-苯亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5- 二酮的制备(化合物3)
所述化合物3的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮(150mg,0.51mmol),苯甲醛(109mg,1.03mmol),无水硫酸镁(210mg),DMF(5mL),碳酸铯(420mg,1.29mmol)置于反应瓶中,氮气保护,45℃反应21小时,TLC检测反应完全。将反应液加入到冷水中,有黄色固体析出,过滤,滤饼用无水乙醇和乙酸乙酯的混合溶剂溶解,滤去不溶物,减压浓缩至干,得黄色固体63mg即为所述化合物3,收率36.65%。
1H NMR(400MHz,DMSO-d6)δ12.31(s,1H),12.22(s,1H),10.00(s,1H),7.84(s,1H),7.52(d,J=8Hz,2H),7.39(t,J=8Hz,2H),7.32(t,J=8Hz,1H),6.73(s,1H),1.37(s,9H);MS(ESI)m/z 338.1715(M+H)+(calcd forC19H20DN4O2338.1722)。
实施例6
(3Z,6Z)-3-苯基氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮的制备(化合物4)
所述化合物4的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(290mg,1mmol),苯氘甲醛(214mg,2mmol),无水硫酸镁(900mg),DMF(8mL),碳酸铯(261mg,0.8mmol)置于反应瓶中,氮气保护,45℃反应至完全。将反应液加入到冷水中,用二氯甲烷提取,减压浓缩至干,得黄色固体183mg即为所述化合物4,收率为54.43%。
1H NMR(400MHz,DMSO-d6)δ12.33(s,1H),12.25(s,1H),10.05(s,1H),7.85(s,1H),7.53(d,J=4Hz,2H),7.43(t,J=8Hz,2H),7.32(t,J=8Hz,1H),6.85(s,1H),1.38(s,9H);MS(ESI)m/z 338.1715(M+H)+(calcd forC19H20DN4O2338.1722)。
实施例7
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮的制备(化合物5)
所述化合物5的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(300mg,1.03mmol),2,3,4,5,6-五氘代苯甲醛(230mg,2.07mmol),DMF(14mL),碳酸铯(505mg,1.55mmol)置于反应瓶中, 氮气保护,于80℃反应6小时,TLC跟踪反应至反应完全。将反应液倒入冷水中,有黄色固体析出。过滤,滤饼用无水乙醇和二氯甲烷的混合溶剂溶解,滤去不溶物,减压浓缩至干,得黄色固体229mg即为所述化合物5,收率50.22%。
1H NMR(400MHz,CDCl3)δ12.40(s,1H),9.33(s,1H),8.11(s,1H),7.60(s,1H),7.03(m,2H),1.47(s,9H);MS(ESI)m/z 342.1984(M+H)+(calcd forC19H16D5N4O2342.1973)。
实施例8
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯基)氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮的制备(化合物6)
所述化合物6的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(290mg,1mmol),2,3,4,5,6-五氘代苯基氘甲醛(224mg,2mmol),无水硫酸镁(900mg),DMF(10mL),碳酸铯(261mg,0.8mmol)置于反应瓶中,氮气保护,45℃反应24小时,至反应完全(TLC检测)。将反应液倒入到冷水中,有黄色固体析出。用二氯甲烷提取,减压浓缩至干,得黄色固体240mg即为所述化合物6,收率70.17%。
1H NMR(400MHz,DMSO-d6)δ12.33(s,1H),12.25(s,1H),10.04(s,1H),7.85(s,1H),6.85(s,1H),1.38(s,9H);MS(ESI)m/z 342.2033(M+ H)+(calcd forC19H15D6N4O2343.2036)。
实施例9
(3Z,6Z)-3-(2,3,4,5,6-五氘代苯)氘代亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮的制备(化合物7)
所述化合物7的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮(250mg,0.86mmol),2,3,4,5,6-五氘代苯氘甲醛(191mg,1.72mmol),无水硫酸镁(350mg),DMF(8mL),碳酸铯(420mg,1.29mmol)置于反应瓶中,置换氮气保护,45℃反应24小时,TLC检测反应完全。将反应液加入到冷水中,有黄色固体析出,过滤,滤饼用无水乙醇和乙酸乙酯的混合溶剂溶解,滤去不溶物,减压浓缩至干,黄色固体138mg即为所述化合物7,收率47%。
1H NMR(400MHz,DMSO-d6)δ12.30(s,1H),12.22(s,1H),10.00(s,1H),7.83(s,1H),1.37(s,9H);MS(ESI)m/z 344.2096(M+H)+(calcd forC19H14D7N4O2344.2098)。
实施例10
(3Z,6Z)-3-(4-氟-2,3,5,6-四氘代苯基)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮的制备(化合物8)
所述化合物8的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(300mg,1.03mmol),4-氟-2,3,5,6-四氘代苯甲醛(265mg,2.07mmol),DMF(6mL),碳酸铯(505mg,1.55mmol)置于反应瓶中,氮气保护,于80℃反应5小时,TLC检测反应完全。将反应液倒入冷水中,有黄色固体析出。过滤,滤饼用无水乙醇和二氯甲烷的混合溶剂溶解,滤去不溶物,减压浓缩至干,得黄色固体231mg即为所述化合物8,收率62.6%。
1H NMR(400MHz,DMSO-d6)δ12.36(s,1H),9.14(s,1H),7.99(s,1H),7.60(s,1H),7.02(s,1H),6.97(s,1H),1.47(s,9H);13C NMR(400MHz,DMSO-d6)δ170.76,162.93,160.97,158.04,156.74,140.76,134.78,131.48,129.98,127.01,124.23,113.25,105.48,32.35,31.08ppm;MS(ESI)m/z359.1818(M+H)+(calcd forC19H16D4FN4O2359.1816)。
实施例11
(3Z,6Z)-3-(4-氟-2,3,5,6-四氘代苯基)亚甲基-6-((5-叔丁基-1H-咪唑-4-基)氘代亚甲基)哌嗪-2,5-二酮的制备(化合物9)
所述化合物9的结构式如下:
其制备方法为:将(Z)-1-乙酰基-3-((5-叔丁基-1H-咪唑-4-基)亚甲基)哌嗪-2,5-二酮(250mg,0.86mmol),4-氟-2,3,5,6-四氘代苯甲醛(220mg,1.72mmol),无水硫酸镁(350mg),DMF(8mL),碳酸铯(420mg,1.29mmol)置于反应瓶中,氮气保护,45℃反应24小时,TLC检测反应完全。将反应液倒入冷水中,有黄色固体析出。过滤,滤饼用无水乙醇和乙酸乙酯的混合溶剂溶解,滤去不溶物,减压浓缩至干,得黄色固体144mg即为所述化合物9,收率为46.59%。
1H NMR(400MHz,DMSO-d6)δ12.30(s,1H),12.22(s,1H),10.11(s,1H),7.83(s,1H),6.72(s,1H),1.37(s,9H);MS(ESI)m/z 360.1886(M+H)+(calcd forC19H15D5FN4O2360.1879)。
表1 实施例3-11获得化合物的结构式和化学名称
实施例12:体外血浆代谢稳定性实验
Plinabulin(脱氢苯基阿夕斯丁)10μmol/mL溶液的配制:取1mg/mL溶液吸取10μL,加入990μL的甲醇水(1:1)稀释为10μg/mL溶液,取10μg/mL溶液337.3μL,加入甲醇水(1:1)溶液662.7μL,配置为1mL的浓度为10μmol/mL的脱氢苯基阿夕斯丁溶液,实验避光操作。
Wistar大鼠血浆的制备:取200g的大鼠,心脏取血,肝素钠抗凝,8000r/min,4℃下离心10min,取血浆。
孵育体系200μL,对应96孔板,依次加入180μL的血浆和20μL的10μmol/L的测试样品。样品混均匀后放入37℃的水浴震荡器中孵育,分别于0,5,10,20,40,60,90,120min加入300μL的含内标的乙腈终止液(浓度为100ng/mL的普萘洛尔的乙腈溶液)。反应混合溶液14000rpm,4℃离心15min,小心吸取上清液至进样小瓶中,LC-MS检测每个化合物的剩余量,结果如图1。图1的实验结果表明,合成的氘代系列化合物在血浆中具有与对照脱氢苯基阿夕斯丁相近的稳定性。
实施例13:体外肝微粒体代谢稳定性实验
溶液的配制(1)K2HPO4缓冲液的配制:K2HPO45.7055g,500mL三蒸水溶解,用HCl调节pH至7.4,浓度为50mmol/L。(2)大鼠的肝微粒体(浓度为20mg/mL)--孵育终浓度为0.5mg/mL;(3)待测试化合物标准溶液的配制—反应缓冲液稀释至2μM,每孔加入100μL,终浓度为1μM.(4)NADPH的配制:称取NADPH 10.42mg,加入2.5mL K2HPO4缓冲液溶解,浓度为5mmol·L-1。(5)内标普萘洛尔乙腈溶液的配制:普萘洛尔2.20mg甲醇溶解,配制成浓度为1mg·mL-1的储备液,用乙腈稀释成100ng·mL-1,实验避光操作。
孵育实验:总体积1mL,含微粒体25μL(由20mg·mL-1稀释至0.5mg·mL-1),NADPH 200μL(由5mmol·L-1稀释至1mmol·L-1),待测化合物溶液20μL(由50μmol·L-1稀释至1μmol·L-1),加入K2HPO4缓冲液755μL至1mL。
各孵育体系置于37℃水浴振荡器中孵育,分别在反应0、5、10、20、40、60、90、120、150、2120、270min从体系中取出50μL,加入100μL含100ng·mL-1普萘洛尔的乙腈终止反应。每组平行三个样品,将各终止反应样品离心(4℃,14000rpm)15min,取上清LC-MS/MS检测每一个化合物及的剩余量,结果见图2,图2实验结果表明,新合成的化合物1、2、3、4、7在体外肝微粒体具有与对照脱氢苯基阿夕斯丁相近的代谢稳定性。
实施例14:细胞分析方法
通过MTT分析方法测定所选化合物对肿瘤细胞A431和Jurkat生长的抑制率。
实验材料:
1)细胞株:人皮肤癌细胞株A431和人急性T细胞白血病细胞Jurkat。细胞生长于RPMI-1640培养基和高糖DMEM(Gibco BRL,Rockville,MD,USA)+10%胎牛血清(含2mM L-glutamine,penicillin and streptomycin)中,置37℃(5%CO2–95%air)孵箱中生长,以0.25%的胰蛋白酶消化传代,1~2d换液1次。
2)化合物的溶解与稀释:根据化合物的分子量和质量,分别用DMSO(SIGMA)配制成10mM的母液,再以RPMI-1640培养基稀释至需要浓 度100μM(DMSO含量≤3‰)。稀释后的药物-20℃保存。
3)RPMI 1640细胞培养基,Gibco公司;胎牛血清,Hyclone公司;细胞消化液,0.25%Trypsin+0.02%EDTA
4)MTT液,MTT干粉(Sigma),用PBS充分溶解配成5mg/ml,0.22μm微孔滤膜过滤后分装,-20℃保存。
实验步骤:
1)细胞接种:取对数生长期的细胞,用胰酶消化后分散成单个细胞,计数,将细胞悬液调整至8.0×104个/mL,每孔180μL接种于96孔板中,使接种的细胞数量为4.0×103个/孔。细胞贴壁后,分别加入含有不同浓度化合物的新鲜培养基,使化合浓度分别是12.5nM、6.25nM,每个浓度设3个复孔,操作时避光,并调整各浓度所含DMSO含量一致。
2)药物孵育72h后,每孔加入20μL MTT(3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide,Sigma;5mg/mL),置孵箱孵育4h后去上清,每孔加150μL DMSO溶解,用酶标仪测定在570nm下的吸光度值。根据公式:抑制率=[(A570对照孔-A570给药孔)/A570对照孔]×100%,计算各浓度的抑制率,结果见表2。
表2 化合物在不同药物浓度下对2种人体细胞株的肿瘤抑制率
注:A431-人皮肤癌细胞株;Jurkat人急性T细胞白血病细胞。Plinabulin(脱氢苯基阿夕斯丁)为阳性对照。
表2实验结果表明,上述化合物1、2、3、4、5、6、7均具有与阳性对照Plinabulin(脱氢苯基阿夕斯丁)接近的抑制肿瘤的活性,其中化合物1、2、3在6.25nM浓度下对A431和Jurkat细胞株的抑制率高于Plinabulin(脱氢苯基阿夕斯丁),说明本发明制得的化合物具有明显的抑制肿瘤的活性,尤其对于皮肤癌和急性T细胞白血病。
实施例15:高内涵成像分析系统
一、实验方案
以Plinabulin(脱氢苯基阿夕斯丁)为阳性对照,设置浓度为12.5nM。根据前期数据筛选出3种样品,分别是化合物1、2、3。所有样品设置浓度为12.5nM。根据特异性免疫荧光染色(Alexa 488)的实验方法,通过高内涵成像分析系统检测所选化合物1、2、3对人表皮癌A431细胞内微管蛋白表达的影响。
实验材料:
1)细胞株:人表皮癌细胞株A431
2)培养基:高糖DMEM+10%胎牛血清
3)Trypsin:胰蛋白酶粉末+乙二胺四乙酸二钠(EDTA)
4)多聚甲醛:分析纯
5)Triton X-100:纯度>98%
6)Tubulin抗体:Mouse,Santa Cruz Biotechnology
7)DyLight 488-羊抗鼠IgG
实验步骤:
1)取对数生长期细胞,用胰酶消化后分散成单个细胞,计数,以1.2×104个/孔接种于96孔板,置于37℃、5%CO2培养箱中培养24h,细胞贴壁。
2)细胞贴壁后,实验组每孔加入含有不同浓度化合物的新鲜培养基,使化合浓度为12.5nM,每个浓度设3个复孔,对照组则加入等倍稀释的溶剂DMSO,选取Plinabulin(脱氢苯基阿夕斯丁)为阳性药,每组设3个复孔,37℃,5%CO2饱和湿度的培养箱中培养24h。
3)24h后吸去培养基,PBS洗3次;4%多聚甲醛固定30min,吸去多聚甲醛,PBS洗3次;0.3%Triton X-100,5%BSA封闭30min,吸去,PBS洗3次;加入Tubulin抗体(鼠抗,1:200稀释),4℃过夜;吸去,PBS洗3次;加入DyLight 488-羊抗鼠IgG(1:1000稀释),室温放置90min
4)高内涵成像分析系统拍照分析。
二、检测结果
检测结果如图3。空白组和DMSO对照组对细胞无影响,细胞骨架组成部分微管蛋白呈发散状均匀分布在细胞中,经Alexa488染色后,可观察到绿色荧光均匀分布于细胞内。
阳性对照Plinabulin(脱氢苯基阿夕斯丁)是已知的能够作用于微管蛋白的小分子化合物,细胞内Alexa488染色的微管蛋白荧光强度明显增强,微管蛋白出现集聚现象,细胞形态也发生明显变化(图3)。样品化合物1、2、3作用A431细胞24h后,细胞内Alexa488染色的微管蛋白荧光强度明显增强,微管蛋白出现集聚现象,细胞形态也发生明显变化。经Hommeny软件进行数据统计,结果如图4所示,与空白和对照组相比,样品化合物1、2、3作用后的Alexa488荧光强度明显增高,与阳性对照相比,样品化合物1、2、3效果高与阳性对照。
通过本发明提供的制备方法制得了一系列氘代脱氢苯基阿夕斯丁类化合物,进一步进行上述血浆代谢稳定性实验、体外肝微粒体代谢稳定性实验、对肿瘤细胞的抑制率实验以及对肿瘤细胞内微管蛋白表达的影响等实验,通过实验结果可以证明本发明所述的氘代脱氢苯基阿夕斯丁类化合物对皮肤癌和白血病细胞具有明显的抑制作用,而且还具有良好的代谢稳定性,所以可以将其用于制备治疗癌症的药物,具有良好的市场应用前景。
以上实施例仅用以说明本发明的技术方案,而非对其进行限制;尽管参照前述实例对本发明进行了详细的说明,对本领域的普通技术人员来说,依然可以对前述实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或替换,并不使相应技术方案的本质脱离本发明所要求保护的技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!