从中药夏天无中制备普罗托品及其药物制剂的方法与流程
本发明涉及中药制药技术领域,特别是涉及从中药夏天无中制备普罗托品及其药物制剂的方法。
背景技术:
夏天无为罂粟科植物紫堇属伏生[Carydolis decumbens(Thunb.)Pers.]的干燥块茎,具有通络止痛的作用,可用于高血压偏瘫、坐骨神经痛、风湿关节炎等。夏天无中含有普罗托品(原阿片碱,Protopine)、四氢巴马亭、巴马亭碱、延胡索乙素、球紫堇碱、山缘草定碱、别隐品碱、药根碱、空褐鳞碱、藤荷包牡丹定碱等多种生物碱。其中,普罗托品(protopine,Pro)又名原阿片碱、原鸦片碱、普罗托平、原托品碱、普鲁托品、前鸦片碱、金英花碱、蓝堇碱、普托品、前陶品,为无色棱柱形或斜方形片状结晶,熔点209~210℃,分子式C20H19NO5,结构式如式(I)所示。普罗托品具有镇痛、解痉、止咳、平喘、改善微血管循环、抗血小板聚集,促进戊巴比妥钠的催眠作用,并有弱的杀菌和抗肿瘤作用。可用于治疗眼底充血及抗血栓等。自20世纪7O年代起,国内外学者对其药理活性及机制进行了研究,在临床上有很好的应用前景,但至今未见其开发为临床药品。
迄今为止,国内外对夏天无化学成分研究较多,但对夏天无中普罗托品的提取、分离的报道较少,对夏天无中普罗托品产业化的提取、分离更少报道。现有技术公开了一种从植物中提取普罗托品的方法,该方法存在以下缺限与不足:①该方法中普罗托品制备的原药材泛指含普罗托品的植物药材,而含普罗托品的植物药材较多,其中如含与普罗托品理化性质相同或相似的成分,则难以将普罗托品分离纯化;②该普罗托品制备的方法条件下得到的产品得率较低,且溶剂未进行回收,成本较高,不适用于工业化生产;另有一现有技术公开了从夏天无中提取普罗托品的方法,该方法也存在以下缺限与不足:该方法采用氯仿和甲醇对生物碱中的普罗托品进行结晶,由于氯仿和甲醇的沸点相近,氯 仿难以先行挥发干尽,而氯仿对普罗托品有较大的溶解度,因而普罗托品难以结晶或结晶量少,得率较低,且制备过程中的溶剂也未进行回收,成本较高,不适用于工业化生产。
技术实现要素:
基于此,有必要提供一种从中药夏天无中制备普罗托品的方法。
一种从中药夏天无中制备普罗托品的方法,包括如下步骤:
(1)夏天无醇浓缩液的提取
将夏天无药材粉碎,以体积浓度75-85%的乙醇溶液(优选为体积浓度85%)或以酸调pH 2-3后的体积浓度75-85%的乙醇溶液回流提取,提取液过滤浓缩后,得夏天无醇浓缩液;
(2)夏天无总生物碱的分离
于所述夏天无醇浓缩液中加入酸溶液调PH值为2-3,搅拌溶解(45-50℃加热溶解或超声溶解)后,静置过滤,滤液用碱溶液调pH值10-11,静置过滤,滤渣干燥(离心脱水或50℃干燥)后得夏天无总生物碱;
(3)总生物碱中普罗托品的结晶
于所述夏天无总生物碱中加入氯仿或水饱和氯仿,与乙醇的混合液,或二氯甲烷,搅拌溶解后,过滤,滤液在搅拌下加入体积浓度为85-95%的乙醇溶液,然后加热至40-50℃并回收二氯甲烷至尽,或加热至65-75℃并回收氯仿至尽,余下的溶液冷却后静置至结晶完全,过滤或倾去上清液,得普罗托品结晶,其中,所述混合液中乙醇的体积占比为0-20%;所述混合液或二氯甲烷,与体积浓度为85-95%的乙醇溶液的体积比为1:(0.5-2);
以所述普罗托品结晶重复或不重复步骤(3)的结晶步骤,即得普罗托品。
在其中一个实施例中,步骤(3)所述混合液或二氯甲烷,与体积浓度为 85-95%的乙醇溶液的体积比为1:(0.6-0.9)。
在其中一个实施例中,步骤(3)所述混合液中乙醇的体积占比为1-6%。
在其中一个实施例中,步骤(3)所述加热回收氯仿至尽的加热温度为70-73℃。
在其中一个实施例中,步骤(1)所述夏天无药材粉碎的方法为将夏天无药材粉碎成5-10目的粗粉。将夏天无药材粉碎合理控制在本申请的粒度,可以获得较好提取率,同时便于工业操作,若粒度过大,则提取率会较低;粒度过小,则容易糊化阻止溶媒的流动交换,也会影响溶媒提取率,而且难以过滤,影响生产。
在其中一个实施例中,步骤(1)所述回流提取的方法为分别以原料8-10倍重量的所述乙醇溶液提取2-3次,每次1.5-2h。
在其中一个实施例中,步骤(1)所述浓缩的方法为将提取液减压浓缩至相对密度为1.08-1.15g/cm3,并回收乙醇。浓缩后,所述夏天无醇浓缩液的固含量为25-32%。
在其中一个实施例中,步骤(2)所述酸溶液为质量浓度0.8-1%的盐酸水溶液;所述碱溶液为质量浓度5-8%NaOH水溶液。所述盐酸水溶液的质量浓度优选为0.8%,所述NaOH水溶液的质量浓度优选为8%。
本发明还提供一种普罗托品药物制剂,包括上述方法制备得到的普罗托品或所述普罗托品用盐酸、硫酸、氢溴酸或醋酸制备成的盐,以及药学上可接受的辅料。
在其中一个实施例中,所述普罗托品药物制剂的剂型为片剂、丸剂、滴丸、胶囊剂、软胶囊、颗粒剂、注射剂、粉针剂、滴眼液、外用药液或乳膏。
本发明的原理及优点如下:
本发明在充分研究夏天无药材所含化学成分的理化性质,以及普罗托品的特有的理化性质的基础上,针对现有技术的不足,通过精心实验研究与验证,得出一种从中药夏天无中制备普罗托品的新方法。该方法在保证普罗托品纯度的同时,提高了普罗托品的产率,降低生产成本,提高生产效率,完善了普罗托品工业生产方法,具有工业可操作性,其原理如下:
(1)经过实验对提取参数进行筛选,选用体积浓度75-85%的乙醇溶液作为提取溶剂,可对夏天无中的总生物碱进行较为充分的提取,以保证最终普罗托品的得率,同时,将提取液浓缩至相对密度为1.08-1.15g/cm3的浓缩液,可有效回收提取溶剂,循环利用,降低生产成本;
(2)利用生物碱的酸溶碱沉的原理,将夏天无总生物碱与夏天无醇的浓缩液中的其它成分分离;
(3)本发明经大量的实验研究发现,生物碱类成分中的普罗托品在本发明所选用的二氯甲烷或含醇氯仿(即氯仿或水饱和氯仿与乙醇的混合液,尤其是当该混合液中乙醇的体积占比为1-6%时)中有较好的溶解度,且较难溶于甲醇、乙醇(尤其是含水乙醇)、丙酮、乙醚和醋酸乙酯等,而生物碱中的其它成分在甲醇、乙醇、丙酮、乙醚和醋酸乙酯等中均有较好的溶解性,在此基础之上,本发明将从总生物碱中结晶普罗托品的溶剂选用为二氯甲烷或含醇氯仿与体积浓度为85-95%的乙醇的配合,并进一步确定其体积比为1:(0.5-2),以获得最佳的普罗托品得率。
此外,体积浓度85-95%的乙醇沸点相对较高,通过适当温度(二氯甲烷为40-50℃,氯仿为65-75℃)的加热,可使沸点相对较低的二氯甲烷或氯仿从混合液中较为彻底的分离,回收完全,降低成本的同时,普罗托品因二氯甲烷或氯仿的挥发回收而难溶于未挥发的含水乙醇溶液结晶析出,进一步有效提高普罗托品的得率,而其它生物碱成分因在未挥发的乙醇溶液中有较好的溶解性则保留在含水乙醇溶液中,可实现通过溶剂置换结晶分离法达到普罗托品的分离纯化之目的。
与现有技术相比,本发明具有以下有益效果:
(1)本发明总生物碱的提取选用了通过方法优选后的含水乙醇溶液,可回收循环使用,不仅不污染环境,而且可大量节省生产成本。
(2)普罗托品的分离与纯化选用了通过方法优选后少量的二氯甲烷或含醇氯仿与特定体积浓度的含水乙醇溶液按照一定比例配合,在分离与纯化过程中,二氯甲烷或含醇氯仿在较低温度下回收,特别是二氯甲烷在此工艺中的首次使用,低温回收的溶剂可循环使用,降低成本,同时产品得率高、节省能耗,而 且减少了环境污染。
(3)本发明结晶普罗托品使用溶媒少:只使用乙醇与二氯甲烷或乙醇与氯仿二种溶媒,且溶媒可回收循环使用;用时短、成本低:不需用柱分离与复杂的溶媒系统,常用生产设备即可生产;普罗托品的得率高、纯度高:本发明生产普罗托品得率可高达0.52%,纯度可高达100%。
(4)采用本发明所提供的方法,工艺流程简单,操作方便,产品质量控制方便,有可操作性,是适合于工业生产的安全可靠的制备方法。
附图说明
图1为夏天无药材原料粉碎粒度与普罗托品的提取量曲线图;
图2为夏天无醇浓缩液提取正交效应曲线图;
图3普罗托品分离工艺的正交实验效应曲线图。
具体实施方式
以下结合具体实施例对本发明的从中药夏天无中制备普罗托品及其药物制剂的方法作进一步详细的说明。
实施例1 夏天无醇浓缩液的提取
1、仪器与试药
1.1仪器:DZLW-s-b型电电热恒温水浴锅:北京市永光明医疗仪器有限公司;RE-5210A型旋转蒸发器:上海亚荣生化仪器厂;SHZ-95A型循环水式多用真空泵:巩义市予华仪器有限责任公司;Waters 2695-2998型高效液相色谱仪;Melter Toledo XP 26型电子天平;Sartorius BL310型电子天平;Sartorius BS223s型电子天平;Precisa 3100C型电子天平;东方-B型风热式电热恒温干燥箱;Q-168型中药参茸切片机:上海冰都电器有限公司;CDE-260双杯搅拌机:广州市嘉发家电器厂;HYD-04A型高速多功能粉粹机:中山市好运达电器有限公司。
1.2试药:夏天无药材(由广东省采芝林有限公司提供),经鉴定为罂粟科植物紫堇属伏生[Carydolis decumbens(Thunb.)Pers.]的干燥块茎;普罗托品(原阿 片碱,供含量测定用,购自中国药品生物制品检定所,批号110853-200402);乙腈(色谱纯,购于赛默飞世尔科技(中国)有限公司);其它试剂均为分析纯。
2、普罗托品含量测定方法
高效液相色谱法(RP-HPLC),采用中国药典2010年版一部夏天无含量测定项下的方法。
2.1色谱条件与系统适用性试验
色谱柱:Agilent Eclipse plus C18(4.6mm×150mm,5μm);Agilent Eclipse XPB-C18(4.6mm×150mm,5μm);以乙腈-三乙胺醋酸溶液(取三乙胺8ml,冰醋酸30ml,加水稀释至1000ml)(18:82)为流动相,流速:1mL·min-1;柱温:25℃;检测波长:289nm。
2.2对照品溶液的制备
精密称取普罗托品(原阿片碱)对照品1mg,精密称定,置25ml量瓶中,加1%的盐酸溶液0.5ml使溶解,加甲醇至刻度,摇匀,作为对照品溶液。
2.3供试品溶液的制备
按各样品的制备工艺进行提取,备用。
3、夏天无药材原料普罗托品提取最佳粒度研究
3.1实验目的
夏天无药材为干燥块茎,质地较坚实,若直接用溶媒提取,普罗托品难以溶出,需将夏天无药材进行粉碎。粉碎的粒度对溶媒对普罗托品的提取率有较大的影响,粉碎的粒度过大,则提取率会较低;粉碎的粒度过小,则容易糊化阻止溶媒的流动交换,也会影响溶媒提取率,而且难以过滤,因此,夏天无药材需要有一个适宜的粉碎粒度。
本实验的目的:探讨夏天无药材中提取普罗托品的原料最佳粉碎粒度。
3.2夏天无药材原料最佳粉碎粒度选择试验
等量称取夏天无原料10份各120g,第1份不粉碎,其余9份用粉碎机分别粉碎成2目、10目、12目、18目、24目、50目、60目、80目、100目的粉末,分别准确称取80g,分别加入800ml的85%的乙醇溶液,相同条件下水浴98℃加热回流提取1.5h取出,相同条件下趁热过滤(350目滤布),冷却放置过夜, 量取每份滤液的体积量,取滤液作为供试品溶液,用上述RP-HPLC法测定每份滤液中普罗托品的含量,并计算每份样品中被提取的普罗托品的含量。见表1。
表1夏天无药材原料粉碎粒度与提取量
以夏天无药材原料粉碎粒度为横坐标,以样品中被提取的普罗托品的提取量为纵坐标作图,见图1。
3.3结果与结论
从表1与图1可以看出,普罗托品的提取量与夏天无药材原料粉碎粒度存在一定的对应关系,普罗托品的提取量随粉碎粒度的增大而增高,但当粉碎粒度达到5-10目以后普罗托品提取量基本保持稳定。因此,实验发现:从夏天无药材原料提取普罗托品的最佳粒度为5-10目。结论:优选的夏天无药材原料最佳粉碎粒度为5-10目,且适合于工业化生产。
4、含普罗托品的夏天无醇浓缩液最佳提取工艺优选
4.1实验目的
筛选含普罗托品的夏天无醇浓缩液最佳提取方法。
在本试验过程中,我们以不同体积浓度的乙醇为提取溶剂,不同提取时间,不同提取次数和溶剂用量为因素,用L9(34)正交设计,以普罗托品含量,提取液 的得膏率为指标,选择最佳的提取工艺。
4.2正交实验法优选夏天无提取工艺
4.2.1夏天无提取工艺的正交实验设计
选用L9(34)因素水平表,结合成分性质及生产实际选择乙醇浓度,乙醇用量,提取时间,提取次数为因素,各因素水平见表2。根据正交安排9种提取工艺,见表3。
表2提取条件因素水平表
4.2.2夏天无提取工艺的正交实验方法与结果
等量称取9份样品各160g,以表3的9种工艺组合进行回流提取。
表3提取条件正交试验表
各提取液趁热过滤(300目滤布),冷却放置过夜,量取每份滤液的体积量,取滤液作为供试品溶液,用上述RP-HPLC法测定每份滤液中普罗托品的含量,并计算每份样品中被提取的普罗托品的含量,以上9份提取液经减压浓缩得到 不含或少含乙醇的流浸膏,测定各流浸膏的干固物含量,计算得膏率。干固物测定方法:用胶头滴管取约1g流浸膏,置干燥至恒重的蒸发皿中,精密称定,于烘箱中105℃干燥至恒重,由剩余的重量和取样量计算流浸膏的干固物含量。见表4。
表4.含普罗托品的夏天无醇浓缩液提取正交实验结果
4.2.3夏天无提取工艺的正交实验分析
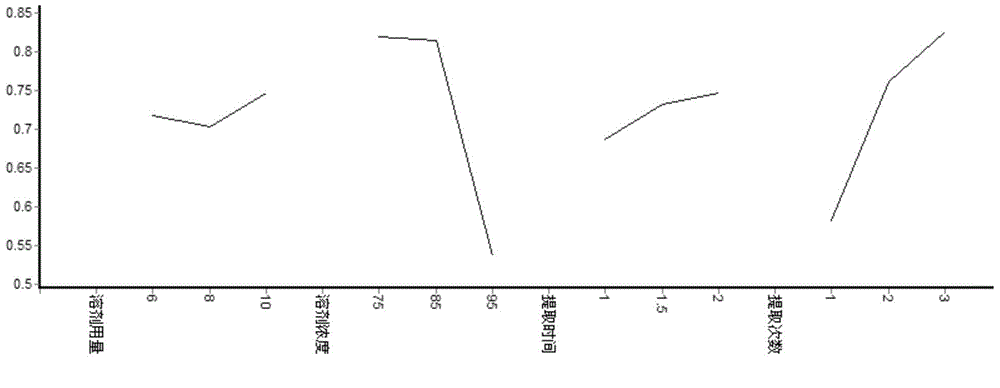
由含普罗托品的夏天无醇浓缩液提取正交直观分析(表5)、含普罗托品的夏天无醇浓缩液提取正交效应曲线图(图2)和含普罗托品的夏天无醇浓缩液提取正交方差分析(表6),可知各因素对夏天无提取工艺有一定影响,各因素作用主次为B>D>C>A,即影响因素由大到小的顺序为:溶剂浓度、提取次数、提取时间、溶剂用量。综合分析,其最佳优化提取工艺为:A3B2C3D3,即加10倍量85%乙醇提取3次,每次回流提取2h。
表5含普罗托品的夏天无醇浓缩液提取正交直观分析
注:原阿片碱评分=(原阿片碱含量/原药材取样量)×0.98;干膏收率评分=(得膏率/原药材取样量)×0.02;综合评分=原阿片碱评分+干膏收率评分。
表6含普罗托品的夏天无醇浓缩液提取正交方差分析
4.2.3夏天无最佳提取生产工艺的优选
从夏天无提取工艺的正交实验分析可知:各因素作用主次为B>D>C>A,即影响因素由大到小的顺序为:溶剂浓度、提取次数、提取时间、溶剂用量。综合分析的最佳优化提取工艺为A3B2C3D3,即加10倍量85%乙醇提取3次,每次回流提取2h。但从方差分析表中可知:影响因素之间无显著性差异,从生产节能与节约成本等多方面考虑,综合最佳优提取工艺条件:A3B2C3D3,拟定优选夏天无最佳提取生产工艺条件:A3B2C3D1+A2B2C2D1,即以85%乙醇提取2次,第一次加10倍量,回流提取2h,第二次加8倍量,回流提取1.5h。
4.3夏天无最佳提取生产工艺验证
为了考察夏天无最佳优化提取工艺(A3B2C3D3即加10倍量85%乙醇提取3 次,每次回流提取2h)与最佳提取生产工艺(A3B2C3D1+A2B2C2D1,即加85%乙醇提取2次,第一次加10倍量,回流提取2h,第二次加8倍量,回流提取1.5h)的合理性,对夏天无最佳提取生产工艺进行验证。取夏天无800g,共2份,分别按正交试验优选的最佳条件A3B2C3D3回流提取与拟定的最佳提取生产工艺条件A3B2C3D1+A2B2C2D1回流提取,收集醇提液;照前述测定方法测定普罗托品,以验证夏天无最佳提取生产工艺。验证结果见表7。
表7工艺验证样品中普罗托品含量
从表7可知,拟定的夏天无最佳提取生产工艺条件A3B2C3D1+A2B2C2D1提取的提取液中普罗托品的总量与夏天无正交最佳工艺条件A3B2C3D3提取的提取液中普罗托品含量相近,而拟定的夏天无最佳提取生产工艺条件既减少了乙醇的使用量,又节省了时间与能耗。说明拟定的夏天无最佳提取生产工艺条件:A3B2C3D1+A2B2C2D1即以85%乙醇提取2次,第一次加10倍量,回流提取2h,第二次加8倍量,回流提取1.5h可行。
实施例2 夏天无总生物碱的分离
1、仪器与试药
1.1仪器:DZLW-s-b型电电热恒温水浴锅:北京市永光明医疗仪器有限公司;RE-5210A型旋转蒸发器:上海亚荣生化仪器厂;SHZ-95A型循环水式多用真空泵:巩义市予华仪器有限责任公司;Waters 2695-2998型高效液相色谱仪;Melter Toledo XP 26型电子天平;Sartorius BL310型电子天平;Sartorius BS223s型电子天平;Precisa 3100C型电子天平;东方-B型风热式电热恒温干燥箱;Q-168型中药参茸切片机:上海冰都电器有限公司;CDE-260双杯搅拌机:广州市嘉发家电器厂;HYD-04A型高速多功能粉粹机:中山市好运达电器有限公司。 1.2试药:夏天无药材(由广东省采芝林有限公司提供),经鉴定为罂粟科植物紫堇属伏生[Carydolis decumbens(Thunb.)Pers.]的干燥块茎;普罗托品(原阿片碱,供含量测定用,购自中国药品生物制品检定所,批号110853-200402);乙腈(色谱纯,购于赛默飞世尔科技(中国)有限公司);其它试剂均为分析纯。
2、普罗托品含量测定方法
高效液相色谱法(RP-HPLC),采用中国药典2010年版一部夏天无含量测定项下的方法。
2.1色谱条件与系统适用性试验
色谱柱:Agilent Eclipse plus C18(4.6mm×150mm,5μm);Agilent Eclipse XPB-C18(4.6mm×150mm,5μm);以乙腈-三乙胺醋酸溶液(取三乙胺8ml,冰醋酸30ml,加水稀释至1000ml)(18:82)为流动相,流速:1mL·min-1;柱温:25℃;检测波长:289nm。
2.2对照品溶液的制备
精密称取普罗托品(原阿片碱)对照品1mg,精密称定,置25ml量瓶中,加1%的盐酸溶液0.5ml使溶解,加甲醇至刻度,摇匀,作为对照品溶液。
2.3供试品溶液的制备
按各样品的制备工艺进行提取,备用。
3、含普罗托品的夏天无总生物碱的分离
3.1实验目的
探索从含普罗托品的夏天无醇浓缩液中分离含普罗托品的夏天无总生物碱的条件方法。在本试验过程中,我们以不同质量浓度的盐酸为酸溶试剂,以不同质量浓度的氢氧化钠为碱沉试剂,用上述HPLC法测定夏天无总生物碱中普罗托品的含量,优化从含普罗托品的夏天无醇浓缩液中分离含普罗托品的夏天无总生物碱的工艺方法。
3.2实验方法
取按实施例1中最佳提取生产工艺条件提取的夏天无醇浓缩液,测定相对密度,用质量浓度0.8%或1.0%的盐酸溶液调pH值2-3,于50℃加热溶解,放冷,静置12小时以上,过滤。取滤液,用质量浓度5%或8%的NaOH溶液调pH值10-11, 静置12小时以上,抽滤,沉淀加水洗涤1-2次,减压抽至无水滴流出,得夏天无总生物碱湿品,称重,50℃减压真空干燥,得夏天无总生物碱干品,称重。测定夏天无总生物碱中普罗托品的含量。
3.3实验结果与结论
实验结果见表8-9。结果表明:用上述二种浓度的HCl酸化试剂与二种浓度的NaOH碱化试剂,通过酸溶碱沉法从夏天无醇浓缩液中分离含普罗托品的夏天无总生物碱,结果所用试剂量、所得总生物碱重量及总生物碱中所含普罗托品量均保持较稳定,说明二种浓度的HCl酸化试剂与二种浓度的NaOH碱化试剂均较好适用于含普罗托品的夏天无总生物碱的分离。从能将普罗托品溶解更完全考量,故生产选用0.8%的HCl酸化试剂进行酸化溶解;从能将普罗托品沉淀更完全考量,故生产选用8%的NaOH碱化试剂进行碱化沉。
表8.含普罗托品的夏天无总生物碱的分离(一)
表9.含普罗托品的夏天无总生物碱的分离(二)
结论:含普罗托品的夏天无总生物碱的分离条件选定为:取夏天无醇浓缩液,按1g密度为1.08-1.15g/cm3的流浸膏加0.8%盐酸溶液2-5ml的比例,45-50℃加热或超声溶解,调pH值2-3,放冷,静置12小时以上,过滤。取滤液,用8%的NaOH溶液调pH值10.0-11.0,静置12小时以上,过滤,沉淀加水洗涤2次,离 心脱水或50℃干燥,得含普罗托品的夏天无总生物碱。
实施例3 夏天无总生物碱中普罗托品的分离与纯化
1、仪器与试药
1.1仪器:DZLW-s-b型电电热恒温水浴锅:北京市永光明医疗仪器有限公司;RE-5210A型旋转蒸发器:上海亚荣生化仪器厂;SHZ-95A型循环水式多用真空泵:巩义市予华仪器有限责任公司;Waters 2695-2998型高效液相色谱仪;Melter Toledo XP 26型电子天平;Sartorius BL310型电子天平;Sartorius BS223s型电子天平;Precisa 3100C型电子天平;东方-B型风热式电热恒温干燥箱;Q-168型中药参茸切片机:上海冰都电器有限公司;CDE-260双杯搅拌机:广州市嘉发家电器厂;HYD-04A型高速多功能粉粹机:中山市好运达电器有限公司。
1.2试药:夏天无药材(由广东省采芝林有限公司提供),经鉴定为罂粟科植物紫堇属伏生[Carydolis decumbens(Thunb.)Pers.]的干燥块茎;普罗托品(原阿片碱,供含量测定用,购自中国药品生物制品检定所,批号110853-200402);乙腈(色谱纯,购于赛默飞世尔科技(中国)有限公司);其它试剂均为分析纯。
2、普罗托品含量测定方法
高效液相色谱法(RP-HPLC),采用中国药典2010年版一部夏天无含量测定项下的方法。
2.1色谱条件与系统适用性试验
色谱柱:Agilent Eclipse plus C18(4.6mm×150mm,5μm);Agilent EclipseXPB-C18(4.6mm×150mm,5μm);以乙腈-三乙胺醋酸溶液(取三乙胺8ml,冰醋酸30ml,加水稀释至1000ml)(18:82)为流动相,流速:1mL·min-1;柱温:25℃;检测波长:289nm。
2.2对照品溶液的制备
精密称取普罗托品(原阿片碱)对照品1mg,精密称定,置25ml量瓶中,加1%的盐酸溶液0.5ml使溶解,加甲醇至刻度,摇匀,作为对照品溶液。
2.3供试品溶液的制备
按各样品的制备工艺进行提取,备用。
3、夏天无总生物碱中普罗托品的分离
3.1实验目的
从夏天无总生物碱中分离普罗托品,建立一种简单可行,稳定性高的分离普罗托品的方法。在本试验过程中,我们以不同体积浓度的乙醇为结晶溶剂、不同倍量的二氯甲烷或含醇氯仿(所述含醇氯仿中乙醇的体积占比为1-6%)、不同倍量的含水乙醇、不同结晶温度为因素,用L9(34)正交设计,以普罗托品含量,普罗托品的结晶量,普罗托品得率为指标,选择最佳的分离工艺。
3.2正交实验法优选普罗托品的分离工艺
3.2.1优选普罗托品分离工艺的正交实验设计
选用L9(34)因素水平表,结合成分性质及生产实际选择含醇氯仿(以含醇氯仿为例,二氯甲烷类似)用量、乙醇用量、乙醇浓度、回收温度为因素,各因素水平见表10。根据正交安排9种分离工艺,见表11。
表10优选普罗托品分离条件因素水平表
表11优选普罗托品分离分离条件正交试验表
3.2.2优选普罗托品分离工艺的正交实验方法与结果
分别称取9份夏天无总生物碱各8g,按表11所列的9种工艺组合,分别加入含醇氯仿,于50℃水浴加热溶解,过滤,滤渣加少量含醇氯仿洗涤1-2次,洗涤液滤过,并入滤液中,滤液分别加入9种工艺组合相应量与浓度的乙醇,分别按正交表的9种工艺加热温度加热蒸馏回收混合溶液中的氯仿,回收至所加入的氯仿完全蒸出,剩余含水乙醇混合溶液体积略少于加入的含水乙醇的体积时,停止回收,回收含醇氯仿液再生后循环使用;剩余含总生物碱的含水乙醇混合溶液放冷,5-25℃下静置24-120小时析晶。待结晶相对完全后,滤取结晶,晾干,称重,普罗托品粗晶。用上述HPLC法测定每份普罗托品粗晶中普罗托品的含量,结果见表12。
表12优选普罗托品分离正交实验结果
3.2.3普罗托品分离工艺的正交实验分析
由普罗托品分离工艺的正交实验直观分析(表13)、普罗托品分离工艺的正交效应曲线图(图3)和普罗托品分离工艺的正交实验方差分析(表14)可知,各因素对普罗托品分离工艺有一定影响,各因素作用主次为D>A>C>B,即影响因素由大到小的顺序为:回收温度、氯仿用量、乙醇浓度、乙醇用量。综合分析,其最佳优化分离工艺为:A3B1C1D3,即用16倍量含醇氯仿溶解总生物碱,加入10倍量75%乙醇,75℃水浴加热蒸馏回收混合溶液中的氯仿。
表13普罗托品分离工艺的正交实验直观分析
注:普罗托品析晶量评分=析晶量×0.3;普罗托品粗晶含量评分=Pro含量×0.7;综合评分=普罗托品析晶量评分+普罗托品粗晶含量评分
表14普罗托品分离工艺的正交方差分析
3.2.4普罗托品分离工艺的优选
由普罗托品分离工艺的正交实验分析可知,各因素对夏天无总生物碱中分离普罗托品有一定的影响,各因素作用主次为D>A>C>B,即影响因素由大到小 的顺序为:回收温度、氯仿用量、乙醇浓度、乙醇用量,最佳优化分离工艺为:A3B1C1D3,即用16倍量含醇氯仿溶解总生物碱,加入10倍量75%乙醇,75℃水浴加热蒸馏回收混合溶液中的氯仿。但从方差分析表中可知:影响因素之间无显著性差异,从实验过程中发现,浓度为85%以下的乙醇,在加入氯仿溶液过程中会出现混浊分层现象,给操作带来不便;含醇氯仿的用量12倍与16倍对结果几乎不产生影响,即用12倍量含醇氯仿可将总生物碱溶解完全;由此,最佳分离工艺优选为:A2B2C1D3,即用12倍量含醇氯仿溶解总生物碱,加入10倍量85%乙醇,75℃水浴加热蒸馏回收混合溶液中的氯仿。在加热蒸馏回收混合溶液中的氯仿过程中发现,比较适宜的加热蒸馏回收温度为73℃;当氯仿回收待尽时,宜将加热蒸馏回收温度调整为70℃,当在此温度下加热蒸馏回收几无液体蒸出时,混合溶液中的氯仿回收接尽完全。综合考虑,最终确定优化工艺为:用12倍量含醇氯仿溶解总生物碱,加入10倍量的85-95%乙醇,73℃加热蒸馏回收混合溶液中的氯仿,当氯仿回收待尽时,将加热蒸馏回收温度调整为70℃,继续加热蒸馏回收几无液体蒸出时,停止回收,溶液放冷,静置至结晶完全,过滤或倾去上清液,得含生物碱普罗托品的粗品。
3.3普罗托品优选分离工艺验证
为了考察普罗托品优选分离工艺的合理性,取夏天无总生物碱约8g,精确称定,分别按正交试验的最佳条件:A2B2C1D3(即用12倍量含醇氯仿溶解总生物碱,加入10倍量85%乙醇,75℃水浴加热蒸馏回收混合溶液中的氯仿)与最终确定的普罗托品生产优选工艺条件(即用12倍量含醇氯仿溶解总生物碱,加入10倍量的85%或95%乙醇,73℃加热蒸馏回收混合溶液中的氯仿,当氯仿回收待尽时,将加热蒸馏回收温度调整为70℃,继续加热蒸馏回收几无液体蒸出时,停止回收。)溶液放冷,静置至结晶完全,过滤或倾去上清液,得含生物碱普罗托品的粗品。晾干,称重,用上述HPLC法测定普罗托品粗品中普罗托品的含量。结果见表15。
表15普罗托品优选分离工艺验证
3.4结果与讨论
从表15普罗托品优选分离工艺验证中可以看出,普罗托品分离的优选工艺与正交最佳工艺相比,得到的普罗托品粗晶量与粗晶中普罗托品含量均保持稳定,且相近,而优选工艺易于控制,更适合于工业化生产,说明优选工艺可行。
4、普罗托品的纯化
4.1普罗托品的纯化方法
普罗托品的纯化是采用上述优化工艺,对普罗托品粗晶进行重结晶,将普罗托品粗结晶,置于回流提取装置中,按1g普罗托品粗结晶加入12ml含醇氯仿的比例,50℃加热回流溶解20-30min,放冷,过滤。不溶物加少量含醇氯仿洗涤2次,洗涤液并入滤液中,在搅拌状态下缓缓加入85%与95%的乙醇溶液,使含醇氯仿液体积:85%或95%的乙醇溶液体积=1:0.82,水浴加热器于73℃加热蒸馏回收混合溶液中的氯仿,当氯仿回收待尽时,将加热蒸馏回收温度调整为70℃,继续加热蒸馏回收几无液体蒸出时,停止回收,溶液放冷,静置至结晶完全,过滤,得含生物碱普罗托品的精品。晾干,称重,用上述HPLC法测定普罗托品精品中普罗托品的含量。
4.2普罗托品的纯化结果
通过上述普罗托品的纯化方法纯化,得含生物碱普罗托品98%以上的普罗托品结晶精品。结果见表16。
表16普罗托品的纯化
结果表明:上述普罗托品的纯化方法稳定可行。
实施例4 普罗托品的制备一
取夏天无药材(批号:20140812,按药典方法测得普罗托品含量为0.72%)1600g,粉碎成8目的粗粉,用体积浓度85%乙醇溶液回流提取2次,第一次加入10倍量85%乙醇,回流提取2h,过滤,药渣再加入8倍量85%乙醇,回流提取1.5h过滤,合并两次滤液,减压回收乙醇,回收至浓缩液中不含乙醇,测得夏天无醇浓缩液的相对密度为1.125g/cm3(50℃热测),干固物含量为26.85%,得重量为1487.5g的夏天无醇浓缩液,计算含夏天无醇浓缩液的浸膏量为:干固物含量为26.85%×天无醇浓缩液重量1487.5g=399.4g。
取上述夏天无醇提浓缩液,加入质量浓度0.8%盐酸820ml,50℃水浴加热,搅拌,测得pH值为2.04,放冷,静置过夜,过滤。滤液在搅拌状态下缓缓加入质量浓度8%的NaOH溶液200ml,测得pH值为10.45,静置沉淀,过夜,减压抽滤,沉淀加100ml水洗涤2次,减压抽干,得总生物碱湿品91.22g,50℃烘干,得总生物碱干品22.81g。
取上述夏天无总生物碱干品,加入274ml氯仿与乙醇的混合液(其中乙醇的体积占比为1.0%),50℃水浴加热溶解,过滤。不溶物,加20ml氯仿与乙醇的混合液洗涤2次,并入滤液中,取滤液,加入230ml的体积浓度85%乙醇,73℃水浴加热蒸馏回收混合溶液中的氯仿,当氯仿回收待尽时(混合溶液体积接近230ml),将加热蒸馏回收温度调整为70℃,继续加热蒸馏回收几无液体蒸出时(混合溶液体积接近220ml),停止回收,溶液放冷,静置120小时,至结晶完全,滤取结晶,晾干,得含生物碱普罗托品的粗晶8.92g。用HPLC法测得普罗托品粗晶中普罗托品的含量为95.43%。
取上述普罗托品粗晶,加入135ml氯仿与乙醇的混合液(其中乙醇体积占比1.5%),50℃水浴加热溶解,过滤,不溶物加10ml氯仿与乙醇的混合液洗涤2次,并入滤液中,取滤液,加入100ml的体积浓度95%乙醇,73℃水浴加热蒸馏回收混合溶液中的氯仿,当氯仿回收待尽时(混合溶液体积接近120ml),将加热蒸馏回收温度调整为70℃,继续加热蒸馏回收几无液体蒸出时(混合溶液体积接近95ml),停止回收,溶液放冷。静置72小时,至结晶完全,滤取结晶,晾干,得 含生物碱普罗托品的精品8.31g。用HPLC法测得普罗托品精品中普罗托品的含量为99.87%。得率为0.52%。
对比例1
另取夏天无药材(批号:20140812,按药典方法测得普罗托品含量为0.72%)1600g,按现有技术(专利申请号为200410042521.X,一种从植物中提取原阿片碱及其药物制剂的制备和应用)的方法制备普罗托品,结果得普罗托品的精品2.48g,普罗托品的含量为98.92%,得率为0.16%。其结果与实施例4结果比较分析如下:
1、提取所消耗乙醇量比较与分析:二种方法的乙醇消耗量比较见表17。本发明使用乙醇量、乙醇浓度等是通过优化后的条件,使用的乙醇浓度相对较低,用量相对较少,并由于提取次数少、提取时间短,不仅节约了能耗,而且乙醇的消耗量明显少于现技术的量。
表17提取所消耗乙醇量比较
2、分离所消耗氯仿的比较与分析:二种方法的氯仿消耗量比较见表18。本发明使用的是含醇氯仿,且是通过优化后的条件,使用的含醇氯仿因少量的乙醇存在增加了对普罗托品的溶解度,氯仿用量相对较少,并由于蒸馏回收温度相对较高,所需的回收时间短,约1.5小时。而现技术的方法由于蒸馏回收温度相对较低(60℃),未能达到氯仿混合液蒸馏回收时的沸腾温度,故与本发明蒸馏回收1.5小时时的氯仿回收量比较,回收氯仿量明显少,因此本发明消耗氯仿量大大少于现技术的量,减少了对环境的污染。
表18分离所消耗氯仿的比较
3、普罗托品的得率与转移率的比较与分析:二种方法的普罗托品得率与转移率的比较见表19。从表19中可见,本发明所得普罗托品不仅纯度高,而且得率与转移率都很高。而现技术的方法所得普罗托品除纯度较高外,得率与转移率都较低,其技术缺限有二:其一,现技术的方法由于分离与重结晶时所加的氯仿:乙醇的体积比=1:3-5,远高于而本发明所加的含醇氯仿:85%乙醇的体积比=1:0.5-2中的乙醇,蒸馏回收氯仿后,现技术的方法剩余的乙醇量远高于本发明的方法剩余的乙醇量,而在剩余的乙醇溶液中,由于其它成分的助溶作用,乙醇溶液对普罗托品也有一定的溶解作用,其乙醇量越大溶解的普罗托品越多,所得的普罗托品结晶量越少。其二,现技术的方法蒸馏回收温度相对较低(60℃),未能达到氯仿混合液蒸馏回收时的沸腾温度,难以蒸馏回收氯仿,蒸馏回收混合液中的275ml氯仿,约需要48小时,而本发明只需约1.5小时;因此现技术的方法不仅耗时、耗能,而且难以产业化,而本发明则相反。
表19普罗托品得率与转移率的比较
综上所述,本发明的方法与现有技术的方法相比不仅能减少环境污染、减少能耗、节约成本,而且所得普罗托品纯度高、得率与转移率都很高,符合产业化要求。
实施例5 普罗托品的制备二
取夏天无药材(批号:20140923,按药典方法测得普罗托品含量为0.42%)1600g,粉碎成8目的粗粉,用含0.5%盐酸的体积浓度75%乙醇溶液(pH 2-3)回 流提取2次,第一次加入10倍量含0.5%盐酸的体积浓度75%乙醇溶液,回流提取2h,过滤,药渣再加入8倍量含0.5%盐酸的体积浓度75%乙醇溶液回流提取1.5h,过滤。合并两次滤液,减压回收乙醇,回收至浓缩液中不含乙醇,测得浓缩液的相对密度为1.128g/cm3(53℃热测),干固物含量为31.14%,得重量为1623.5g的夏天无酸性醇提浓缩液。
取上述夏天无酸性醇提浓缩液。加入质量浓度0.8%盐酸21ml,50℃水浴加热,搅拌,测得pH值为2.13,放冷,静置过夜,过滤。滤液在搅拌状态下缓缓加入质量浓度8%的NaOH溶液210ml,测得pH值为10.65,静置沉淀,过夜,减压抽滤,沉淀加100ml水洗涤2次,减压抽干,得总生物碱湿品90.15g。
取上述夏天无总生物碱湿品,加入270ml二氯甲烷,室温搅拌溶解,过滤。不溶物,加20ml二氯甲烷洗涤2次,并入滤液中,取滤液,加入220ml的体积浓度95%乙醇,45℃水浴加热蒸馏回收混合溶液中的二氯甲烷,当二氯甲烷回收待尽时(加热蒸馏回收几无液体蒸出,混合溶液体积接近220ml),停止回收,溶液放冷。静置48小时,至结晶完全,滤取结晶,晾干,得含生物碱普罗托品的粗晶4.98g。用HPLC法测得普罗托品粗晶中普罗托品的含量为93.34%。
取上述普罗托品粗晶,加入75ml二氯甲烷,室温搅拌溶解,过滤。不溶物,加10ml二氯甲烷洗涤2次,并入滤液中,取滤液,加入75ml的体积浓度95%乙醇,45℃水浴加热蒸馏回收混合溶液中的二氯甲烷,当二氯甲烷回收待尽时(加热蒸馏回收几无液体蒸出,混合溶液体积接近75ml),停止回收,溶液放冷。静置48小时,至结晶完全,滤取结晶,晾干,得普罗托品的粗晶4.63g。用HPLC法测得普罗托品精品中普罗托品的含量为100.34%。得率为0.29%。
对比例2
另取夏天无药材(批号:201400923,按药典方法测得普罗托品含量为0.42%)1600g,按现有技术(专利申请号为200410042521.X,一种从植物中提取原阿片碱及其药物制剂的制备和应用)的方法制备普罗托品,结果得普罗托品的精品1.92g,普罗托品的含量为99.13%,得率为0.12%。其结果与实施例5结果比较分析如下:
本实施例的发明技术与现有技术的方法的比较,和实施例4与现有技术的方法的比较基本相同,与实施例4不同的是:①本实施例中的夏天无药材的批号与普罗托品含量有所不同;②本实施例中的分离与重结晶时所用的溶媒有所不同。本实施例中使用二氯甲烷替代含醇氯仿,起到了含醇氯仿相同的效果,但毒性对环境的影响却减少了十倍(氯仿的毒性为二氯甲烷的十倍);二氯甲烷作为普罗托品分离与纯化的关键溶媒,在本发明中为首先使用。本实施例中本发明与现有技术所得普罗托品的得率与转移率的比较见表20。
表20普罗托品得率与转移率的比较
从表20中可见,本发明所得普罗托品不仅纯度高,而且得率与转移率也都很高。而现技术的方法所得普罗托品除纯度较高外,得率与转移率都较低。
综上所述,本发明的方法与现有技术的方法相比,本发明不仅创新使用了二氯甲烷作为关键性溶媒、减少了环境污染、减少了能耗、节约了成本,而且提高了所得普罗托品纯度、得率与转移率,符合产业化要求。
实施例6 普罗托品(实施例4制备得到)片剂、胶囊剂的制备
一、普罗托品片剂的制备
取普罗托品90g,粉碎成粉末,加入淀粉900g、阿拉伯胶4g、羟丙基甲基纤维素钠3g,混匀,喷洒95%食用乙醇20ml制成软材,置制粒机内制成颗粒(一号筛大小),于60℃下烘干至含水份量3-5%,经上述干燥整粒后,加入润滑剂硬脂酸镁,充分混合均匀,上压片机压制成0.25g/片,装瓶,经检验合格后,包 装即得成品。
二、普罗托品胶囊剂制备
普罗托品 80g
可溶淀粉 920g
取普罗托品80g,粉碎成粉末,加入可溶淀粉920g混匀,喷洒95%食用乙醇20ml制成软材,置制粒机内制成颗粒(一号筛大小),于60℃下烘干至含水份量3-5%,用一号筛整粒,装3号胶囊,装瓶,经检验合格后,包装即得成品。
实施例7 普罗托品(实施例5制备得到)注射剂的制备
一、普罗托品水针剂的制备
普罗托品 10.0g
氯化钠 20.0g
甘露醇 50.0g
取普罗托品10.0g,在100级以下车间内,加入氯化钠20.0g、甘露醇50.0g、注射用水9000,充分搅匀后,加入1%的盐酸水溶液适量,50℃加热使溶解,调节PH值3.0-5.0,搅匀,加注射用水调整总体积至10000ml,加入0.1%的活性碳80℃保温15min,过滤,滤液经0.2μm微孔滤膜超滤,得澄明溶液,溶液无菌分装于安瓶中,每安瓶5ml,溶封,灭菌,经检验合格后,包装即得成品。
二、普罗托品粉针剂的制备
普罗托品 10.0g
氯化钠 20.0g
甘露醇 50.0g
取普罗托品10.0g,在100级以下车间内,加入氯化钠20.0g、甘露醇50.0g、注射用水9000,充分搅匀后,加入1%的盐酸水溶液适量,50℃加热使溶解,调节PH值3.0-5.0,搅匀,加注射用水调整总体积至10000ml,加入0.1%的活性碳80℃保温15min,过滤,滤液经0.2μm微孔滤膜超滤,得澄明溶液,溶液无菌分装于10ml西林瓶中,每瓶5ml,加通气的内盖,经冷冻干燥,轧盖,灭菌。经检验合 格后,包装即得成品。
实施例8 普罗托品(实施例4制备得到)滴眼液的制备
取普罗托品5.0g,在100级以下车间内,加入氯化钠90.0g、尼泊金甲酯0.23g、尼泊金丙酯0.11g蒸馏水100,充分搅匀后,加入1%的盐酸水溶液适量,50℃加热使溶解,调节PH值3.0-5.0,搅匀,加蒸馏水调整总体积至1000ml,加入0.1%的活性碳80℃保温15min,过滤,滤液经0.2μm微孔滤膜超滤,得澄明溶液,溶液无菌灌装于滴眼瓶中,每瓶2ml,100℃灭菌30min,经检验合格后,包装即得成品。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明/构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!