柯义巴肽A类似物及其合成方法与应用与流程
本发明是涉及柯义巴肽A类似物及其合成方法与应用,属于固相有机合成领域。
背景技术:
US20120028905A1公开了McPhail等从巴拿马海洋蓝藻Leptolyngbya sp.中分离纯化得到的一种高度N-甲基化的天然环酯肽—柯义巴肽A(coibamide A),其对肺癌、乳腺癌、黑色素瘤、白血病以及中枢神经癌等都具有低纳摩尔的细胞毒性,同时对乳腺、中枢神经以及卵巢癌细胞具有较好的组织选择性。研究表明coibamide A是通过一种新的作用机制来抑制癌细胞增殖,因此,可将coibamide A作为一种具有良好前景的抗癌药物的先导物。然而,由于自然界中存在的coibamide A十分有限,采用从藻类中分离纯化coibamide A需要复杂的工艺,大量的有机溶剂,较长的时间,并且coibamide A的收率很低,无法满足研究以及应用的需求,例如无法满足对coibamide A进行相关药效学评估的需求。
2014年He Wei(Total synthesis of proposed structure of coibamide A,a highly N-and O-methylated cytotoxic marine cyclodepsipeptide.Tetrahedron Lett.2014,55,6109-12)首次报道了一种coibamide A的全合成方法,该方法采用[(4+1)+3+3]的片段拼接策略,通过多步液相反应,合成了coibamide A。然而该合成方法需要多种缩合试剂,耗时长,并且由于在多肽缩合过程中存在氨基酸的消旋现象,因而合成效率很低。
相较于费时的液相合成方法,采用固相多肽合成方法(SPPS)可使coibamide A的合成简便、快速。2015年,Nabika等(Synthesis and biological evaluation of the[D-MeAla11]-epimer of coibamide A.Bio.Med.Chem.Lett.2015,25,302-6.)以TCP树脂为固相载体,通过Fmoc策略合成了包含全部氨基酸单元的环化前体,以TFA/DCM(5:95,v/v)切除树脂后,最后通过在溶液中进行大环内酯化反应合成11位MeAla发生异构化的coibamide A的异构体,在环化反应中,环化收率仅3.8%,以第一个氨基的取代度计算,合成该coibamide A的异构体的总收率仅1%。
综上可知,现有技术提供的coibamide A的合成方法耗时长、收率低,并且由于反应过程中会发生消旋反应,也难以确保所合成的分子为所设计的目标分子。因此开发一种高效、快速合成coibamide A或coibamide A异构体或其类似物的方法,以满足研究以及应用的需求是本领域亟待解决的技术问题。
技术实现要素:
本发明的目的之一在于提供一种柯义巴肽A(coibamide A)类似物的合成方法,该合成方法可快速、高效的合成所设计的柯义巴肽A类似物。
本发明的另一目的在于提供一种柯义巴肽A的异构体,该异构体具有良好的抗细胞过度增殖活性。
本发明的再一目的在于提供所述柯义巴肽A的异构体的应用。
为实现上述目的,一方面,本发明提供一种柯义巴肽A类似物的合成方法,该柯义巴肽A类似物具有式I所示的结构式:
式I中,R1、R2、R3、R5、R7、R8、R9、R11、R13、R14各自独立地为氢、甲基或乙基;
R4为甲氧基苄基、乙氧基苄基、甲胺基苄基或乙胺基苄基;
R6、R10各自独立地为正丁基、异丁基或仲丁基;
R12为甲氧基甲基、乙氧基甲基、甲氧基乙基或乙氧基乙基;
G为氢、甲基、
或氨基保护基;该氨基保护基为Boc、Fmoc、Cbz、Trt或Allyl;
R15、R21、R22各自独立地为正丙基、异丙基、异丁基或仲丁基;
R16、R17、R19、R20、R23、R24各自独立地为氢、甲基、乙基或氨基保护基;
R18为各自独立地为甲氧基甲基、乙氧基甲基、甲氧基乙基或乙氧基乙基;
Y为羟基、甲氧基、氨基、甲氨基或双甲基氨基;
X为O、N或N(CH3);
优选地,式I具有式I-1所示的结构:
更优选地,式I-1具有式I-1-1或式I-1-2所示的结构:
所述方法包括如下步骤:
a.提供肼树脂;
b.采用活化剂,固相有机合成式A~式F中任一种肼树脂负载的环化前体:
式A~式F任一种中的PNH代表氨基保护基;该氨基保护基为Boc、Fmoc、Cbz、Trt或Allyl,优选Fmoc;
c.在氧化剂的存在下进行切割反应,使步骤b中所得肼树脂负载的环化前体中的多肽链与树脂分离,得到多肽链环化前体;
优选地,所述多肽链环化前体的碳末端为羧基;
d.脱除步骤c中所得多肽链环化前体上氮末端上的保护基PNH后,采用缩合剂进行环合反应,得到式I所示化合物。
本发明研究发现步骤b中式A~式F的肼树脂负载的环化前体在切割后,可有效进行环合反应,显著提高合成柯义巴肽A类似物的产率,当将本发明所述的方法应用于合成柯义巴肽A或其立体异构体时,以第一个氨基的取代度计算,其产率可高达8%~20%,远远优于现有技术提供的柯义巴肽A或其立体异构体的合成路线。
优选地,在本发明所述方法的步骤b中,采用活化剂,固相有机合成式D所示的肼树脂负载的环化前体;
更优选地,固相有机合成如式D-1所示的肼树脂负载的环化前体:
进一步优选地,固相有机合成式D-1-1或式D-1-2所示的肼树脂负载的环化前体:
根据本发明的具体实施方案,在本发明所述的方法中,式D-1所示的肼树脂负载的环化前体是按如下方法制备得到:
b-1.采用活化剂,固相有机合成式D-1-F1所示的肼树脂负载的主链:
优选地,固相有机合成式D-1-1-F1或D-1-2-F1所示的肼树脂负载的主链:
式D-1-F1、式D-1-1-F1或式D-1-2-F1中所述POH为羟基保护基,优选叔丁基、三苯甲基、烯丙基或苄基;
b-2.采用去保护基剂脱除步骤b-1所得肼树脂负载的主链上的羟基保护基POH;
b-3.在步骤b-2中暴露出的羟基上进行酯化反应以制得式D-1-F2所示的结构:
脱除式D-1-F2中的氨基保护基PNH后,继续偶联形成式D-1所示的肼树脂负载的环化前体;
优选地,在步骤b-2暴露出的羟基上进行酯化反应以制得式D-1-1-F2或式D-1-2-F2所示的结构:
脱除式D-1-1-F2或式D-1-2-F2的氨基保护基PNH后,继续偶联以形成式D-1-1或式D-1-2所示的肼树脂负载的环化前体。
本发明通过选用特定的树脂以及保护基策略使步骤b-2在脱除POH的情况下不使树脂与肽链的连接键断裂,有效合成了式D-1所示的肼树脂负载的环化前体。
本领域技术人员应当知晓,当采用一系列构型确定的氨基酸时,应用本发明的合成方法可制备得到柯义巴肽A,或某一确定构型的柯义巴肽A异构体或其类似物。例如在步骤b-1中选用一系列构型确定的氨基酸,采用活化剂,固相有机合成式D-1-1-F2,并在步骤b-3中形成式D-1-2,切割关环后可制备现有技术公开的确定构型的式I-1-2所示的柯义巴肽A;又例如在步骤b-1中选用一系列构型确定的氨基酸,采用活性剂,固相有机合成主链式D-1-1-F1,并在步骤b-3中形成式D-1-1,切割并关环后可制备式I-1-1所示的确定构型的柯义巴肽A的异构体。
根据本发明的具体实施方案,在步骤b-1中,采用的试剂为Fmoc-MeIle7-OH、Fmoc-MeSer(Me)6-OH、Fmoc-MeThr(POH)5-OH、Fmoc-MeLeu4-OH、Fmoc-MeSer(Me)3-OH以及Me2Val1-Hiv2-OH;或
采用保护或不保护的选自以下序列中的部分或全部片段为试剂:Me2Val1-Hiv2-MeSer(Me)3-MeLeu4-MeThr(POH)5-MeSer(Me)6-MeIle7。
本领域技术人员应当知晓,当采用的试剂为Fmoc-MeIle7-OH、Fmoc-MeSer(Me)6-OH、Fmoc-MeThr(POH)5-OH、Fmoc-MeLeu4-OH、Fmoc-MeSer(Me)3-OH以及Me2Val1-Hiv2-OH时,实际上是在肼树脂上依次逐一偶联以形成所述的肼树脂负载的主链。当采用的试剂为保护或不保护的选自主链序列中的部分或全部片段为试剂时,实际上是将符合肼树脂负载的主链肽序列的片段依次偶联到树脂上,上述片段的组合形式以及个数不受限制,只要能形成所述式D-1-F1所示的肼树脂负载的主链即可,例如在脱除Fmoc基团的肼树脂上偶联Fmoc-MeIle7-OH后,依次偶联Fmoc-MeThr(POH)5-MeSer(Me)6-OH、Fmoc-MeSer(Me)3-MeLeu4-OH及Me2Val1-Hiv2-OH;又例如在脱除Fmoc基团的肼树脂上偶联Fmoc-MeIle7-OH后,依次偶联Fmoc-MeLeu4-MeThr(POH)5-MeSer(Me)6-OH以及Me2Val1-Hiv2-MeSer(Me)3-OH。
根据本发明的具体实施方案,在步骤b-3脱除式D-1-F2中的氨基保护基PNH之后,采用的试剂为Fmoc或Boc基团保护的Tyr(Me)10、MeLeu9或Ala8;或
在步骤b-3脱除式D-1-F2中的氨基保护基PNH之后,采用Fmoc或Boc基团保护的选自以下序列中的部分或全部片段为试剂:Tyr(Me)10-MeLeu9-Ala8。
本领域技术人员应当知晓,当采用的试剂为Fmoc或Boc基团保护的Tyr(Me)10、MeLeu9或Ala8时,实际上是在脱除氨基保护基PNH的式D-1-1-F2上依次逐一偶联以形成式D-1。当采用的试剂为Fmoc或Boc基团保护的选自Tyr(Me)10-MeLeu9-Ala8序列中的部分或全部片段为试剂时,实际上是将符合该序列的片段依次偶联到脱除氨基保护基PNH的式D-1-1-F2上,上述片段的组合形式以及个数不受限制,只要能形成式D-1即可。
根据本发明的具体实施方案,在步骤b或步骤b-1中,所述的活化剂为DCC、DIC、HATU、HBTU、HCTU、HOAt、HOBt、PyBOP、BOP-Cl、光气、双光气和三光气中的一种或多种的组合,优选光气、双光气或三光气;
更优选地,在步骤b-1中,采用光气、双光气或三光气各自独立地活化待连接的Fmoc基团保护的单体氨基酸或氨基酸片段以及Me2Val1-Hiv2-OH,活化步骤包括:将待连接的Fmoc基团保护的单体氨基酸或氨基酸片段,或Me2Val1-Hiv2-OH和光气、双光气或三光气溶于惰性溶剂中,加入有机碱,反应得到相应的单体氨基酸酰氯或氨基酸片段酰氯,或Me2Val1-Hiv2-Cl,其中,以光气或将双光气、三光气换算成光气后的摩尔数计算,光气的摩尔数为所述Fmoc基团保护的单体氨基酸或氨基酸片段,或Me2Val1-Hiv2-OH的摩尔数的1.0~2.0倍;
更优选地,在步骤b-3中,采用光气、双光气或三光气各自独立地活化待连接的Fmoc或Boc基团保护单体氨基酸或氨基酸片段,活化步骤包括:各自独立地将Fmoc或Boc基团保护单体氨基酸或氨基酸片段和光气、双光气或三光气溶于惰性溶剂中,加入有机碱,反应得到相应的单体氨基酸酰氯或氨基酸片段酰氯,其中,以光气或将双光气、三光气换算成光气后的摩尔数计算,光气的摩尔数为Fmoc或Boc基团保护单体氨基酸或氨基酸片段的摩尔数的1.0~2.0倍;进一步优选地,在步骤b-3中,反应得到相应的单体氨基酸酰氯或氨基酸片段酰氯之后,立即加入HOAt使单体氨基酸酰氯或氨基酸片段酰氯转化成相应的酯;其中,HOAt的摩尔数为Fmoc或Boc基团保护单体氨基酸或氨基酸片段的摩尔数的0.5~1.0倍;
其中,在步骤b-1或步骤b-3中,所述的有机碱为三甲基吡啶、二异丙基乙胺、吡啶、二甲基吡啶和2-甲基喹啉中的一种或多种的组合;
所述的惰性溶剂为四氢呋喃、二噁烷、二甘醇二甲醚和1,3-二氯丙烷中的一种或多种的组合。
本发明研究发现当单体氨基酸为蛋白氨基酸(非N-甲基化氨基酸)时,使用BTC活化所产生的酰氯会发生多种副反应,降低缩合效率,通过加入HOAt将酰氯转化为相应-OAt酯,可显著提高其与空间位阻大、亲核能力差的氨基酸(例如氮原子上带有甲基等取代基的氨基酸)的固相缩合效率,可在相对较短的时间内提高缩合产率。
根据本发明的具体实施方案,在步骤a中,所述肼树脂为苯肼树脂;更优选地,所述肼树脂的替代度为0.1~1.0mmol/g。
根据本发明的具体实施方案,在步骤c中,所述的氧化剂为醋酸铜、N-溴代丁二酰亚胺和氧气中的一种或多种的组合;
在步骤d中,所述的缩合剂为EDC·HCl、DCC、DIC、DMTMM+BF4-、DMTMM+Cl-、HATU、HBTU、HCTU、HOAt、HOBt、BOP、BOP-Cl、PyBOP、PyAOP、DPPA、FDP和FDPP中的一种或多种的组合。
本发明中步骤c中的氧化剂的作用为将肼键氧化成二氮烯键,随后在切割剂的作用下,例如水的作用下使步骤b所得肼树脂负载的环化前体中的多肽链与树脂分离,得到多肽链环化前体,本领域技术人员应该知晓该步可以先进行氧化再切割树脂,也可以在氧化的同时进行树脂的切割。
另一方面,本发明提供一种柯义巴肽A的异构体,该柯义巴肽A的异构体具有式I-1-1所示的结构:
本发明研究发现该柯义巴肽A的异构体具有良好的抗细胞过度增殖的活性。
对比现有技术公开的式I-1-2所示的柯义巴肽A的结构与式I-1-1所示的柯义巴肽A的异构体的结构可以看出,该柯义巴肽A的异构体2位羧酸以及11位氨基酸的构型发生了改变,而由于构型的改变带来了预料不到的抗细胞增殖活性。
再一方面,本发明提供式I-1-1所示的柯义巴肽A的异构体在制备用于治疗过度增殖性疾病的药物中的应用;
优选地,其中所述的过度增殖性疾病包括结肠癌、直肠癌、脑瘤(优选为成胶质细胞瘤)、肺癌(优选为非小细胞肺癌)、表皮鳞癌、膀胱癌、胰腺癌、乳腺癌、卵巢癌、宫颈癌、子宫内膜癌、结直肠癌、肾细胞癌、食管腺癌、食管鳞状细胞癌、非霍奇金淋巴瘤、肝癌、皮肤癌、甲状腺癌、头颈癌、前列腺癌、神经胶质瘤及鼻咽癌中的一种或多种;更优选地,所述的过度增殖性疾病为乳腺癌、非小细胞肺癌。
综上可知,本发明提供一种快速、高效合成所设计的柯义巴肽A类似物的方法,并且利用该方法获得了一种具有良好抗细胞过度增殖的柯义巴肽A的异构体。
本发明中的缩写具有如下定义:
BTC:三光气;EDC·HCl:1-乙基-(3-二甲基氨基丙基)碳酰二亚胺盐酸盐;DCC:N,N'-二环己基碳酰亚胺;DIC:N,N'-二异丙基碳二亚胺;DMTMM+BF4-:4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉四氟硼酸盐;DMTMM+Cl-:4-(4,6-二甲氧基三嗪-2-基)-4-甲基吗啉氯盐;HATU:O-(7-氮苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯;HBTU:苯并三氮唑-N,N,N',N'-四甲基脲六氟磷酸盐;HCTU:6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯;HOAt:1-羟基-7-偶氮苯并三氮唑;HOBt:1-羟基苯并三唑;BOP:苯并三氮唑-1-基氧基三(二甲基氨基)磷鎓六氟磷酸盐;BOP-Cl:双(2-氧代-3-恶唑烷基)次磷酰氯;PyBOP:六氟磷酸苯并三唑-1-基-氧基三吡咯烷基磷;PyAOP:(3H-1,2,3-三唑并[4,5-b]吡啶-3-氧基)三-1-吡咯烷基鏻六氟磷酸盐;DPPA:叠氮磷酸二苯酯;FDP:pentafluorophenyl diphenyl phosphate;FDPP:五氟苯基二苯基磷酸酯;DMAP:4-二甲氨基吡啶;TFA:三氟乙酸;TIS:三异丙基硅烷;Fmoc:9-芴甲氧羰基;Boc:叔丁氧羰基;Cbz(Z):苄氧羰基;Trt:三苯甲基;Allyl:烯丙氧羰基;
Fmoc-MeIle7-OH:Fmoc-(L)-MeIle7-OH:
Fmoc-MeSer(Me)6-OH:Fmoc-(L)-MeSer(Me)6-OH:
Fmoc-MeThr(POH)5-OH:Fmoc-(L)-MeThr(POH)5-OH:
Fmoc-MeLeu4-OH:Fmoc-(L)-MeLeu4-OH:
Fmoc-MeSer(Me)3-OH:Fmoc-(L)-MeSer(Me)3-OH:
Me2Val1—Hiv2-OH:Me2Val1-Hiv2-Cl:
(L)-Me2Val1—(D)-Hiv2-OH:
(L)-Me2Val1—(L)-Hiv2-OH:
Tyr(Me)10:MeLeu9:Ala8:
Tyr(Me)10-MeLeu9-Ala8:
Fmoc-(D)-MeAla11-OH:Fmoc-(L)-MeAla11-OH:
Boc-(L)-Tyr(Me)10-OH:Fmoc-(L)-MeLeu9-OH:
Fmoc-(L)-Ala8-OH:
Me2Val1-Hiv2-MeSer(Me)3-MeLeu4-MeThr(POH)5-MeSer(Me)6-MeIle7:
附图说明
图1为实施例1制备式I-1-1所示的柯义巴肽A的异构体的基本流程图;
图2为实施例1制备苯肼树脂负载的环化前体式4的基本路线图;
图3为实施例1由苯肼树脂负载的环化前体式4制备式I-1-1所示的柯义巴肽A的异构体的基本路线图;
图4为实施例1制备的环化前体6的1H NMR图;
图5为实施例1制备的环化前体6的13C NMR图;
图6为实施例1制备的环化前体6的HRMS图;
图7为实施例1制备的式I-1-1所示的柯义巴肽A的异构体的1H NMR图;
图8为实施例1制备的式I-1-1所示的柯义巴肽A的异构体的13C NMR图;
图9为式I-1-1所示的柯义巴肽A的异构体的HRMS图。
具体实施方式
为了对本发明的技术特征、目的和有益效果有更加清楚的理解,现结合具体实施例及附图对本发明的技术方案进行以下详细说明,应理解这些实例仅用于说明本发明而不用于限制本发明的范围。
实施例1
本实施例提供式I-1-1所示的柯义巴肽A的异构体及其制备方法。
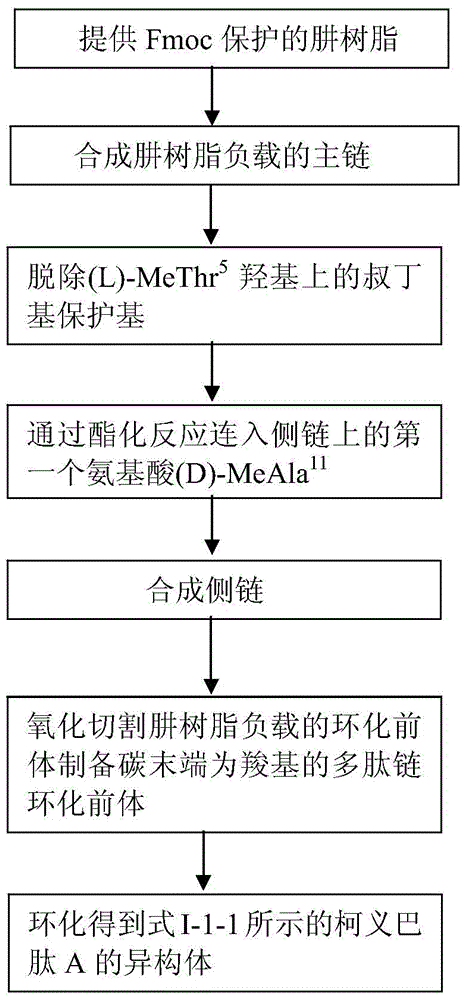
图1大致显示了式I-1-1所示的柯义巴肽A的异构体的制备方法:采用提供Fmoc保护的苯肼树脂;脱除苯肼树脂上的Fmoc保护基后,合成苯肼树脂负载的主链;脱除(L)-MeThr5羟基上的叔丁基保护基;通过酯化反应连入侧链上的第一个氨基酸(D)-MeAla11;合成侧链;氧化切割肼树脂负载的环化前体制备碳末端为羧基的多肽链环化前体;最后环化得到式I-1-1所示的柯义巴肽A的异构体。
具体地,先按图2所示的路线合成苯肼树脂负载的环化前体4。
(1)苯肼树脂负载的主链1的制备:
在10mL的固相反应器中加入以Fmoc保护的苯肼树脂(400mg,0.66mmol/g,0.26mmol,购于Novabiochem)及3mL CH2Cl2,将苯肼树脂溶胀30min,抽除CH2Cl2,用3mL 20%哌啶/DMF溶液脱除苯肼树脂上的Fmoc保护基,10min后,抽除反应液,再次用3mL 20%哌啶/DMF溶液脱除苯肼树脂上的Fmoc保护基,10min后,抽除反应液,采用DMF(4×3mL)洗涤树脂,再用无水THF(2×3mL)洗涤树脂,备用;同时将97mg Fmoc-(L)-MeIle7-OH(0.26mmol)和32mg三光气(BTC,0.11mmol)溶于2mL无水THF,向该溶液中缓慢滴加420μL三甲基吡啶(collidine,3.17mmol),反应立即产生大量白色沉淀,加完后反应3min,将该反应液转移至上述所得脱除Fmoc的苯肼树脂中,再加入870μL DIEA(5.28mmol),N2鼓泡混匀,缩合反应30min后,抽除反应液,用DMF(4×3mL)洗涤树脂,再用3mL无水DMF洗涤树脂;向树脂中加入125μL乙酸酐(1.32mmol)和2mL含有200μL DIEA(1.20mmol)的DMF溶液,反应10min,抽除反应液,用DMF(4×3mL)洗涤树脂,制得Fmoc-(L)-MeIle7-苯肼树脂,其取代度为0.30mmol/g(400mg,0.12mmol,1eq);
如图2所示,重复上述脱保护及缩合步骤,采用逐一偶联的方式,继续依次偶联Fmoc-(L)-MeSer(Me)6-OH、Fmoc-(L)-MeThr(t-Bu)5-OH、Fmoc-(L)-MeLeu4-OH、Fmoc-(L)-MeSer(Me)3-OH以及(L)-Me2Val1—(D)-Hiv2-OH,直至完成苯肼树脂负载的主链1((L)-Me2Val1-(D)-Hiv2-(L)-MeSer(Me)3-(L)-MeLeu4-(L)-MeThr(t-Bu)5-(L)-MeSer(Me)6-(L)-MeIle7-苯肼树脂)的合成,其中脱保护与缩合的步骤基本相同,例如,在制得Fmoc-(L)-MeIle7-苯肼树脂后,用3mL 20%哌啶/DMF溶液脱除Fmoc-(L)-MeIle7-树脂上的Fmoc保护基,10min后,抽除反应液,再次用3mL 20%哌啶/DMF溶液脱除苯肼树脂上的Fmoc保护基,10min后,抽除反应液,用DMF(4×3mL)洗涤树脂,再用无水THF(2×3mL)洗涤树脂,备用。同时活化各待连接的Fmoc-(L)-MeSer(Me)6-OH,具体操作为:将4.0eq的待连接的Fmoc-(L)-MeSer(Me)6-OH和1.64eq的三光气(BTC)溶于2mL无水THF,向该溶液中缓慢滴加12eq三甲基吡啶(collidine),反应立即产生大量白色沉淀,加完反应3min,将该反应液转移到脱除Fmoc保护基的(L)-NHMeIle7-苯肼树脂中,再加入20eq DIEA,N2鼓泡混匀,缩合反应0.5~1h,待四氯苯醌试剂检测至反应完全后,抽除反应液,用DMF(4×3mL)洗涤树脂;
(2)苯肼树脂负载的环化前体4的制备:
将以上制得的苯肼树脂负载的主链1用CH2Cl2(2×3ml)洗涤,抽除CH2Cl2,加入3.0mL TFA/TIS/H2O(v/v/v=95:2.5:2.5)混合溶液脱除叔丁基保护基t-Bu,2min后抽除溶剂,再次加入TFA/TIS/H2O(体积比为95:2.5:2.5)混合溶液(3.0mL)反应5min,用CH2Cl2(2×3ml)及DMF(4×3mL)洗涤树脂,再用3mL无水DMF洗涤树脂,得到脱除t-Bu的苯肼树脂负载的主链2,备用。同时将4.0eq的Fmoc-(D)-MeAla11-OH和2eq DCC溶于2mL CH2Cl2/DMF(v/v=1:1),在0℃下搅拌反应2h,离心分离白色沉淀,将反应液转移到脱除t-Bu的苯肼树脂负载的主链2中,加入催化量的DMAP,N2鼓泡混匀,缩合反应2.5h,抽除反应液,用DMF(4×3mL)洗涤树脂,得到产物3;
用3mL 20%哌啶/DMF溶液脱除3中Fmoc保护基,10min后,用DMF(4×3mL)洗涤树脂,再用无水DMF(2×3mL)洗涤树脂,备用。同时将4.0eq Boc-(L)-Tyr(Me)10-OH和1.64eq三光气(BTC)溶于1mL无水THF,向该溶液中缓慢滴加12eq三甲基吡啶(collidine),反应立即产生大量白色沉淀,向反应体系中加入4.0eq HOAt及1.5mL DIEA/DMF(15%,v/v),反应3min后,将该反应液转移到脱除Fmoc的产物3中,N2鼓泡混匀,缩合反应0.5~1h,待四氯苯醌试剂检测至反应完全后,抽除反应液,用DMF(4×3mL)洗涤树脂;
用CH2Cl2(2×3ml)洗涤树脂,抽除CH2Cl2,加入3.0mL TFA/TIS/H2O(v/v/v=95:2.5:2.5)混合溶液,脱除Boc保护基,2min后抽除溶剂,再次加入3.0mLTFA/TIS/H2O(v/v/v=95:2.5:2.5)混合溶液,反应5min,用CH2Cl2(2×3mL)及DMF(4×3mL)洗涤树脂,再用3mL无水THF洗涤树脂,备用。同时将4.0eq Fmoc-(L)-MeLeu9-OH和1.64eq三光气(BTC)溶于2mL无水THF,向该溶液中缓慢滴加12eq三甲基吡啶(collidine),反应立即产生大量白色沉淀,加完反应3min,将该反应液转移到上述得到的脱除Boc的苯肼树脂中,再加入20eq DIEA,N2鼓泡混匀,缩合反应0.5~1h,待四氯苯醌试剂检测至反应完全后,抽除反应液,用DMF(4×3mL)洗涤树脂;
采用3mL 20%哌啶/DMF溶液脱除上述所得苯肼树脂中的Fmoc保护基,10min后,抽除反应液,再次用3mL 20%哌啶/DMF溶液脱除苯肼树脂上的Fmoc保护基,10min后,抽除反应液,用DMF(4×3mL)洗涤树脂,再用无水DMF(2×3mL)洗涤树脂,备用。同时将4.0eq Fmoc-(L)-Ala8-OH和1.64eq三光气(BTC)溶于1mL无水THF,向该溶液中缓慢滴加12eq三甲基吡啶(collidine),反应立即产生大量白色沉淀,向反应体系中加入4.0eq HOAt及1.5mL DIEA/DMF(15%,v/v),反应3min后,将该反应液转移到脱除Fmoc的苯肼树脂中,N2鼓泡混匀,缩合反应0.5~1h,待四氯苯醌试剂检测至反应完全后,抽除反应液,用DMF(4×3mL)洗涤树脂,完成苯肼树脂负载的环化前体4的合成。
(3)式I-1-1所示的柯义巴肽A的异构体的合成
按图3所示的路线合成式I-1-1所示的柯义巴肽A的异构体:
取出步骤(2)中所得苯肼树脂负载的环化前体4,加入4mL CH2Cl2、10mg Cu(OAc)2、200μL吡啶及200μL水,室温下振荡反应24h后,将树脂滤除,并用20mL CH2Cl2洗涤树脂。将有机相浓缩,残留物用制备型HPLC纯化(10%乙腈—H2O(含1%的TFA)等梯度洗脱5min,10%至100%的乙腈—H2O(含1%的TFA)梯度洗脱25min,保留时间TR=27min),收集产物并将溶剂减压蒸干,得到产物5(27mg,以第一个氨基酸的取代度来计,收率为20%)。HRMS(ESI)m/z:理论计算C80H123N10O19[M+H]+的精确分子量为1527.8960,实测为1527.8963。
在15.3mg产物5(0.01mmol)中加入1.5mL Et2NH与1.5mL CH3CN,室温下搅拌反应15min,减压蒸干溶剂,残留物用制备型HPLC纯化(10%乙腈—H2O(含1%的TFA)等梯度洗脱5min,10%至100%的乙腈—H2O(含1%的TFA)梯度洗脱25min,保留时间TR=20min),收集产物,冷冻干燥后得到白色固体6备用,其1H NMR、13C NMR及HRMS分别如图4、图5及图6所示。
将19.2mg EDC·HCl(0.10mmol)、13.6mg HOAt(0.10mmol)、70μL DIEA(0.40mmol)溶于30mL CH2Cl2中,在冰浴下冷却至0℃。将上述白色固体6溶于CH2Cl2(2mL)中,0℃下缓慢滴加至EDC·HCl/HOAt/DIEA的反应液中,反应1h后撤去冰浴,将反应混合物继续在室温下搅拌反应17h,减压蒸除溶剂,用制备型HPLC纯化(10%乙腈—H2O(含1%的TFA)等梯度洗脱5min,10%至100%的乙腈—H2O(含1%的TFA)梯度洗脱25min,保留时间TR=25min),收集产物,冷冻干燥,得到7.2mg式I-1-1所示的柯义巴肽A的异构体,收率为60%,以第一个氨基酸的取代度来计,总收率为12%,其1H NMR、13C NMR及HRMS分别如图7、图8及图9所示。
[α]D23–58.4°(c 0.10,CHCl3);
IR:3376,2957,1734,1644,1512,1468,1404,1248,1097,757cm-1;
1H NMR(600MHz,CDCl3)δ0.83(d,J=6.6Hz,3H),0.88-0.99(m,18H),1.00(d,J=4.2Hz,3H),1.05-1.08(m,9H),1.11(m,1H),1.11(d,J=7.2Hz,3H),1.28(d,J=6.6Hz,3H),1.31(m,1H),1.36(m,2H),1.50(m,2H),1.60(m,1H),1.68(m,1H),2.02(m,1H),2.05(m,1H),2.21(m,1H),2.34(br s,9H),2.75(s,3H),2.84(m,2H),2.86(s,3H),2.89(m,3H),2.99(m,1H),3.04(s,3H),3.12(s,3H),3.15(s,3H),3.30(s,3H),3.35(s,3H),3.53(m,1H),3.61(m,1H),3.65(dd,J=11.4,4.2Hz,1H),3.75(m,1H),3.77(s,3H),3.83(m,1H),3.90(m,1H),4.73(m,1H),5.00(d,J=5.5Hz,1H),5.11(m,1H),5.32-5.38(m,2H),5.50(br s,1H),5.69(m,1H),6.00(br s,1H),6.35(br s,1H),6.63(br s,1H),6.68(br s,1H),6.77(d,J=9.0Hz,2H),7.09(d,J=8.4Hz,2H);;
13C NMR(100MHz,CDCl3)δ11.6,12.9,15.7,17.9,18.0,18.4,18.6,19.5,19.6,21.2,21.4,23.2,23.3,24.2,24.9,25.3,27.6,28.9,29.7,29.9,30.1,30.2,31.3,32.0,36.2,37.9,38.9,39.4,41.3,47.0,49.9,51.0,51.1,52.5,52.9,55.3,58.6,58.8,63.5,64.7,68.6,69.3,73.8,74.8,113.7,128.4,130.4,158.6,167.2,168.7,169.7,170.0,170.5,171.2,171.3,171.5,172.4ppm;
HRMS(ESI-TOF)m/z:理论计算C65H111N10O16[M+H]+精确分子量为1287.8174,实测为1287.8176。
实施例2
本实施例提供式I-1-2所示的柯义巴肽A及其制备方法。
制备式I-1-2的方法与实施例1制备式I-1-2基本类似,仅以(L)-Me2Val1-(L)-Hiv2-OH替换实施例1中的(L)-Me2Val1-(D)-Hiv2-OH,并以Fmoc-(L)-MeAla11-OH替换实施例1中的Fmoc-(D)-MeAla11-OH,其余步骤均相同,实验过程中以第一个氨基酸的取代度来计,总收率为8%。
[α]D23–128.9°(c 0.7,CHCl3);
IR:2960,2923,1732,1645,1508,1459,1405,1248,1093,1012,756cm-1;
1H NMR(CDCl3,400MHz):δ0.62-1.15(m,36H),1.15-1.38(m,7H),1.40-1.51(m,2H),1.60-1.76(m,2H),2.00-2.10(m,2H),2.12-2.20(m,1H),2.30-2.35(m,6H),2.65-3.40(m,28H),2.45(br s,1H),3.45-3.60(m,2H),3.60-3.70(m,1H),3.70-3.86(m,6H),4.52-5.24(m,5H),5.34-5.60(m,2H),5.60-5.75(m,2H),5.77-5.80(m,0.7H),5.89(d,0.3H,J=4.2Hz),6.57(brs,1H),6.80-6.85(m,2H),6.97(br s,1H),7.10-7.20(m,2H)ppm;
13C NMR(CDCl3,100MHz):δ11.6,13.5,14.1,15.9,18.0,18.1,18.5,18.6,18.7,19.1,19.3,19.6,21.2,21.5,22.7,23.5,23.6,24.2,24.8,25.4,27.7,27.8,28.5,28.8,29.7,29.9,30.1,30.8,30.9,31.0,31.9,32.0,33.4,36.4,38.4,40.0,40.1,41.3,46.5,50.1,50.9,52.1,52.6,55.0,55.1,55.2,55.3,58.7,58.8,58.9,64.1,64.7,68.7,68.9,69.0,74.0,74.1,74.5,74.6,113.9,114.2,114.4,130.2,130.4,158.8,169.6,169.7,170.0,170.1,170.3,170.4,171.6,171.8ppm;
HRMS(ESI-TOF)m/z:理论计算C65H111N10O16[M+H]+的精确分子量为1287.8174,实测为1287.8170。
实施例3式I-1-1所示的柯义巴肽A的异构体的抗细胞过渡增殖活性
1)药物及试剂配制:将1L水加入一袋RPMI1640培养基中,补加2g碳酸氢钠,10万单位青霉素和100mg链霉素,调节pH值至7.4,用0.22μm除菌滤膜过滤除菌。95mL培养基加灭活新生牛血清5mL即为完全培养液。胰蛋白酶用D-hanks缓冲液配成0.25%溶液,过滤除菌后4℃保存备用。
2)待测样品溶液配制:准确称取1.29mg(1.0μmol)实施例1中制备得到的式I-1-1所示的coibamideA的异构体,将其加到灭菌的0.5mL离心管中,加入100μL DMSO,配成10μM原液,-40℃冷冻保存。临用前融化后取适量以完全培养液稀释成相应的浓度。
3)细胞培养及传代:细胞均贴壁培养于含10mL完全培养液细胞培养瓶中,于37℃、5%CO2、饱和湿度下培养。细胞长满瓶底后用灭菌D-hanks液洗两次,加0.25%胰蛋白酶消化细胞2分钟,倒掉胰蛋白酶,待轻摇细胞能完全脱落后,加完全培养液30ml后,用移液管吹散细胞,分装于3个新的细胞培养瓶中,继续培养。
4)药物处理:取刚刚长成完整单层的细胞一瓶,胰蛋白酶消化后收集细胞,用移液管吹打均匀,取两滴细胞悬液台盼蓝(Trypan Blue)染色,于显微镜下计数活细胞数目(死细胞数目不得超过5%),用完全培养液调整细胞数目至1×105个细胞/毫升。于96孔细胞培养板中每孔加入100μL细胞悬液,将培养板置于CO2培养箱中培养12小时,取出培养板后于每孔中补加100μL含不同浓度待测样品的完全培养液,使得药物终浓度分别为1000、100.0、10.0、1.0、0.1和0.01nM,每个浓度设4个平行孔,另设4孔细胞加入不含药完全培养液作正常对照孔。加完药后培养板于微孔板振荡器上振荡混匀,置于CO2培养箱中继续培养72小时。取出培养板,每孔加入5mg/mL的MTT液20μL,振荡混匀,继续培养4小时。弃去每孔中培养液,加100μL的DMSO溶解formazan结晶,振荡10min使完全溶解。
5)细胞毒活性测定:半抑制浓度的用酶标仪测定各孔光吸收,测定波长490nm。根据各孔OD值计算药物对细胞增殖的抑制率:抑制率(%)=[1-OD(加药组)/OD(阴性对照组)]×100%。根据药物浓度的对数对应的抑制率用Microcal Origin软件作直线回归,得到直线方程,计算抑制率在50%时对应的药物浓度即为待测样品对肿瘤细胞的半抑制浓度(IC50),所得数据如表1所示:
表1式I-1-1所示的柯义巴肽A的异构体的IC50值
从表1可以看出本发明所合成的式I-1-1所示的柯义巴肽A具有良好的抗癌细胞活性,尤其是抗乳腺癌细胞及非小细胞癌细胞。
- 还没有人留言评论。精彩留言会获得点赞!