一种帕博西尼晶型A和晶型B的制备方法与流程
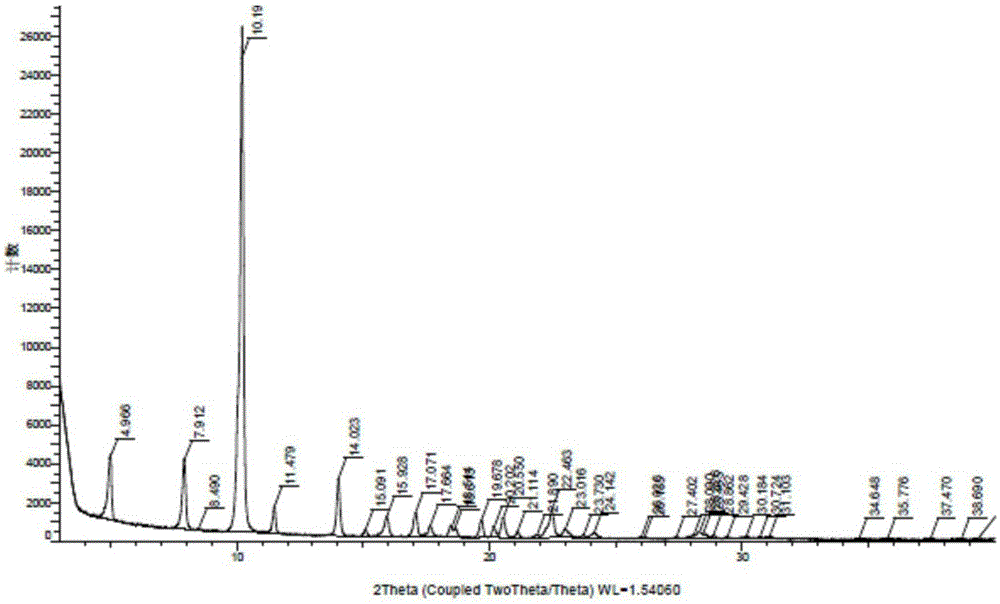
本发明涉及一种6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮的晶型A和晶型B的制备方法,属于化学医药领域。技术背景6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮是由辉瑞开发的一款选择性、每日服用一次的细胞周期蛋白依赖性激酶(CDK)4/6抑制剂,在绝经后妇女ER阳性、HER2阴性形式的晚期乳腺癌中显示出强大疗效,已获FDA突破性药物认证。2015年2月3日,该药物在美国获得了批准上市。WO2014128588A1公开了6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮的游离碱晶型A和晶型B。晶型A的X粉末衍射图在约8.0±0.2、10.1±0.2、10.3±0.2和11.5±0.2度2θ处具有特征峰;晶型B的X粉末衍射图在约6.0±0.2、10.9±0.2、12.8±0.2、16.4±0.2和19.8±0.2度2θ处具有特征峰。考虑到专利中报道的晶型A和晶型B的制备方法,都是采用有机溶剂。而6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮的游离碱形态在所有有机溶剂中溶解度非常差,专利本身给出游离碱的溶剂溶解度信息如下:Kineticsolubilitymeasurementsfor25mg/mLcompound1_freebasesolutionsExperiment#SolventDissolutionTemp.(℃)1n-BuOH>110℃2DMF>110℃3NMP97.94DMSO>110℃5DMAc>110℃6n-Butylacetate>110℃7Anisole>110℃810%n-BuOH/Anisole(v/v)(v/v)>110℃920%n-BuOH/Anisole(v/v)109.71040%n-BuOH/Anisole(v/v)101.41110%n-BuOH/NMP(v/v)103.71225%n-BuOH/NMP(v/v)>110℃1310%1,4-butanediol/anisole(v/v)109.81425%1,4-butanediol/anisole(v/v)104.8151:1:8propyleneglycol/n-BuOH/anisole(v/v)91.2162:1:7propyleneglycol/n-BuOH/anisole(v/v)84.1在高温条件,溶解度只能达25mg/mL。在碱游离时,采用大量有机溶剂高温萃取分液,操作有一定的危险性,污染较大。本发明提供了一种在水或水和可与水混溶的有机溶剂混合溶剂中直接游离得到晶型A和晶型B的方法,工业化生产操作安全便捷,污染较小。技术实现要素:本发明提供了一种如式Ⅰ所示的帕博西尼游离碱晶型A的制备方法,其特征在于包含以下步骤:将帕博西尼游离碱或其盐形式与溶剂中形成混合物,然后将上述溶液用无机碱调碱游离,35~100℃得到帕博西尼游离碱晶型A。所述的盐形式,可以是固体形式,也可以是溶液形式,也可以是单一盐、多种单一盐的混合物或盐与游离碱的混合物。所述的游离碱可以是帕博西尼游离碱的其他固体形式,包括但不限于晶型B或其混晶。所述的盐形式,成盐的酸可以是无机酸,也可以是无机酸。无机酸优选自盐酸、硫酸;有机酸优选自羟乙基磺酸、甲磺酸。所述的无机碱,包括但不限于氨水、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾。所述的溶剂为是水,或水和可与水混溶的有机溶剂混合溶剂。所述可与水混溶的有机溶剂选自甲醇、乙醇、异丙醇、四氢呋喃。本发明的另一目的是提供一种制备帕博西尼游离碱晶型B的制备方法,其特征在于包含以下步骤:将帕博西尼的盐形式溶于溶剂中,然后用无机碱处理调碱游离,在0~20℃得到帕博西尼游离碱晶型B。所述的盐形式,可以是固体形式,也可以是溶液形式,也可以是单一盐、多种单一盐的 混合物或盐与游离碱的混合物。所述的盐形式,成盐的酸可以是无机酸,也可以是无机酸。无机酸优选自盐酸、硫酸;有机酸优选自羟乙基磺酸、甲磺酸。所述的无机碱,包括但不限于氨水、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾。所述的溶剂为水,或水和可与水混溶的有机溶剂混合溶剂。所述可与水混溶的有机溶剂选自甲醇、乙醇、异丙醇、四氢呋喃。附图说明图1为6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮晶型A的XRPD谱图。图2为6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮晶型B的XRPD谱图。具体实施实例:以下将结合实施例对本发明的实施方式做详细说明。本发明的实施方式包括但不局限于下述实施例,其不应当被视为对本发明保护范围的限制。本发明所述的拉帕替尼游离碱晶型A,使用Cu-Kα辐射检测的X射线粉末衍射图谱中,具有以下特征峰,其2θ角度值及相对强度如下表所示:2θ相对强度4.96612.8%7.91214.5%10.193100%11.4795.4%14.02312.1%本发明所述的拉帕替尼游离碱晶型B,使用Cu-Kα辐射检测的X射线粉末衍射图谱中,具有以下特征峰,其2θ角度值及相对强度如下表所示:实施实例1:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮羟乙基磺酸盐的制备取100g4-[6-[(6-(1-丁氧乙烯基)-8-环戊基-7,8-二氢-5-甲基-7-氧代吡啶并[2,3-D]嘧啶-2-基)氨基]-3-吡啶基]-1-哌嗪羧酸叔丁酯于2L三口烧瓶中,加入1.0L甲醇,90.4g羟乙基磺酸,50ml水。加热至60-65℃左右,搅拌3-4小时。将体系降温至0-5℃,析晶,抽滤得黄色固体。将所得固体于甲醇水混合溶剂中重结晶纯化,烘干得黄色蓬松固体94.5g(81.5%收率),HPLC纯度为99.8%。实施实例2:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮盐酸盐的制备取100g4-[6-[(6-(1-丁氧乙烯基)-8-环戊基-7,8-二氢-5-甲基-7-氧代吡啶并[2,3-D]嘧啶-2-基)氨基]-3-吡啶基]-1-哌嗪羧酸叔丁酯于2L三口烧瓶中,加入1.0L甲醇,41.5ml盐酸,100ml水。加热至60-65℃左右,搅拌3-4小时。将体系降温至0-5℃,析晶,抽滤得黄色固体。将所得固体于甲醇水混合溶剂中重结晶纯化,烘干得浅黄色蓬松固体63.4g(72.4%收率),HPLC纯度为99.8%。实施实例3:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮游离碱的制备将20.0g碳酸钠溶于320ml水中搅拌溶解,加热至30℃。将40.0g羟乙基磺酸盐如实施实例1制备)溶于700ml水中,配成溶液,再将此溶液滴入到上述配制的30℃的碳酸钠溶液中,边滴边搅拌,析出黄色固体。滴毕,混合物保温30℃搅拌1小时,抽滤烘干得黄色固体0.97g(97.0%收率),HPLC纯度为99.8%。实施实例4:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将13.1g氢氧化钠溶于1.8L水中搅拌溶解,升温至50℃-60℃。将92.0g羟乙基磺酸盐(如实施实例1制备)溶于1.8L水中,配成溶液,再将此溶液滴入到上述配制的50℃-60℃的氢氧化钠水溶液中,边滴边搅拌,析出黄色固体。滴毕,取样本XRPD监测,已形成结晶形式A。混合物保温50℃-60℃搅拌2小时,抽滤,烘干得黄色固体55.7g(94.7%收率),HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例5:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.43g碳酸钠溶于32ml水中搅拌溶解,加热至35℃。将1.0g游离碱(如实施实例3制备)与0.7g羟乙基磺酸溶于24ml水甲醇混合溶剂中,配成溶液,再将此溶液滴入到上述配制的35℃的碳酸钠溶液中,边滴边搅拌,析出黄色固体。滴毕,取样本XRPD监测,已形成结晶形式A。混合物保温35℃搅拌1小时,抽滤烘干得黄色固体0.97g(97.0%收率),HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例6:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.19g氢氧化钠钠溶于15ml水中搅拌溶解,加热至100℃。将0.80g盐酸盐(如实施实例2制备)溶于20ml水中,配成溶液,再将此溶液滴入到上述配制的100℃的氢氧化钠溶液中,边滴边搅拌,析出黄色固体。滴毕,抽滤烘干得黄色固体0.55g(79.7%收率),HPLC纯度为99.9%,通过XRPD分析为形式A。实施实例7:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.51g碳酸钾溶于32ml水中搅拌溶解,加热至40℃。将1.0g游离碱(如实施实例3制备)与0.3g硫酸溶于25ml水乙醇混合溶剂中,配成溶液,再将此溶液滴入到上述配制的40℃的碳酸钾水溶液中,边滴边搅拌,析出黄色固体。滴毕,保温40℃搅拌1小时,抽滤烘干得黄色固体0.96g(96.0%收率),HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例8:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.21g氢氧化钾溶于30ml水中搅拌溶解,加热至55℃。将1.0g游离碱(如实施实例3制备)与0.45g甲磺酸溶于25ml水异丙醇混合溶剂中,配成溶液,再将此水溶液滴入到上述配制的55℃的氢氧化钾水溶液中,边滴边搅拌,析出黄色固体。滴毕,抽滤,烘干得黄色固体0.95g(95.0%收率),HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例9:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将10g氨水溶于150ml水中搅拌,加热至35℃。将10.0g羟乙基磺酸盐(如实施实例1制备)和盐酸盐(如实施实例2制备)和3g游离碱(式Ⅰ)的混合物溶于250ml水四氢呋喃混合溶剂中,配成溶液,再将此水溶液滴入到上述配制的35℃的氨水溶液中,边滴边搅拌,析出黄色固体。滴毕,保温35℃搅拌0.5小时,抽滤烘干得黄色固体9.1g,HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例10:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式B)的制备将1.0g羟乙基磺酸盐(如实施实例1制备)溶于9ml水中,将溶液缓慢滴入0.22g氢氧化钠与32ml水形成的溶液中,控温20℃,有橙黄色固体析出,取样本XRPD监测,已形成结晶形式B。混合物在20℃,搅拌过夜,抽滤,用水淋洗,烘干得黄色固体0.60g(93.9%收率),通过XRPD分析为形式B。实施实例11:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式B)的制备将0.45g碳酸钠溶于40ml水中搅拌溶解,降温0℃。将1.0g游离碱(如实施实例3制备)与0.7g羟乙基磺酸溶于24ml水甲醇混合溶剂中,配成溶液,再将此溶液滴入到上述配制的0℃的碳酸钠溶液中,边滴边搅拌,析出黄色固体。混合物保温0℃搅拌1小时,抽滤烘干得黄色固体0.97g(97.0%收率),HPLC纯度为99.8%,通过XRPD分析为形式B。实施实例12:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并 [2,3-D]嘧啶-7-酮(形式B)的制备将152.5g羟乙基磺酸盐(如实施实例1制备)和盐酸盐(如实施实例2制备)的混合物溶于1.5L水中,转入5L四口瓶中,将150g氨水与1.5L水混合,于25℃滴入上述混合物中,有大量固体析出,搅拌3小时。抽滤,用水淋洗,烘干得96.1g黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式B。实施实例13:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式B)的制备将0.19g氢氧化钠钠溶于15ml水中搅拌溶解,降温至10℃。将0.80g盐酸盐(如实施实例2制备)和0.3g游离碱(如实施实例3制备)溶于25ml水异丙醇混合溶剂中,配成溶液,再将此溶液滴入到上述配制的10℃的氢氧化钠溶液中,边滴边搅拌,析出黄色固体。滴毕,抽滤烘干得黄色固体0.85g(85.8%收率),HPLC纯度为99.9%,通过XRPD分析为形式B。实施实例14:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式B)的制备将1.0g碳酸钾溶于35ml水中搅拌溶解,加热至15℃。将2.0g游离碱(如实施实例3制备)与0.7g硫酸溶于50ml水乙醇混合溶剂中,配成溶液,再将此溶液滴入到上述配制的15℃的碳酸钾水溶液中,边滴边搅拌,析出黄色固体。滴毕,保温15℃搅拌1小时,抽滤烘干得黄色固体1.85g(92.50%收率),HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例15:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式B)的制备将0.25g氢氧化钾溶于30ml水中搅拌溶解,降温至15℃。将1.0g游离碱(如实施实例3制备)与0.45g甲磺酸溶于25ml水四氢呋喃混合溶剂中,配成溶液,再将此水溶液滴入到上述配制的15℃的氢氧化钾水溶液中,边滴边搅拌,析出黄色固体。滴毕,抽滤,烘干得黄色固体0.94g(94.0%收率),HPLC纯度为99.8%,通过XRPD分析为形式B。实施实例16:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将150g羟乙基磺酸盐(如实施实例1制备)溶于1.5L水中,转入5L四口瓶中,将150g 氨水与1.5L水混合,于25℃滴入上述混合物中,有大量固体析出,搅拌3小时,取样本XRPD监测,已形成结晶形式B。将混合物升温至50-60℃,搅拌4小时,抽滤,用水淋洗,烘干得89.3g(93.2%收率)黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式A。实施实例17:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.5g氢氧化钠溶于20ml水中,加热至100℃,将1.0g形式B(如实施实例12制备)加入其中,搅拌1小时,抽滤,烘干得0.97g(97.0%收率)黄色固体。HPLC纯度为99.8%,通过XRPD分析为形式A。实施实例18:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.5g碳酸钠溶于20ml水和甲醇混合溶剂中,加热至50℃,将1.0g形式B(如实施实例12制备)加入其中,搅拌1小时,抽滤,烘干得0.98g(98.0%收率)黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式A。实施实例19:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将0.5g碳酸钾溶于30ml水和乙醇混合溶剂中,加热至60℃,将1.0g形式B(如实施实例12制备)加入其中,搅拌1小时,抽滤,烘干得0.99g(99.0%收率)黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式A。实施实例18:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮(形式A)的制备将5g氢氧化钾溶于300ml水和异丙醇混合溶剂中,加热至35℃,将10.0g形式B(如实施实例12制备)加入其中,并加入0.2g形式A(如实施实例4制备)作为晶种,搅拌3小时,抽滤,烘干得10.0g(98.0%收率)黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式A。实施实例19:6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并 [2,3-D]嘧啶-7-酮(形式A)的制备将5g氨水溶于300ml水和四氢呋喃混合溶剂中,加热至35℃,将10.0g形式B(如实施实例12制备)与形式A(如实施实例4制备)的混合物加入其中,搅拌3小时,抽滤,烘干得9.9g(99.0%收率)黄色固体。HPLC纯度为99.9%,通过XRPD分析为形式A。本发明提出的一种6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮晶型A和晶型B的制备方法,已通过实施例进行了描述,相关技术人员明显能在不脱离本
发明内容、精神和范围内对本文所述的6-乙酰基-8-环戊基-5-甲基-2-[[5-(哌嗪-1-基)吡啶-2-基]氨基]-8H-吡啶并[2,3-D]嘧啶-7-酮晶型A和晶型B的制备方法进行改动或适当变更与组合,来实现本发明技术。特别需要指出的是,所有相类似的替换和改动对本领域技术人员来说是显而易见的,它们都被视为包括在本发明的精神、范围和内容中。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1