液晶材料中间体以及液晶材料的制备方法与流程
本发明涉及液晶材料领域,特别涉及中间体及液晶材料的制备方法。
背景技术:
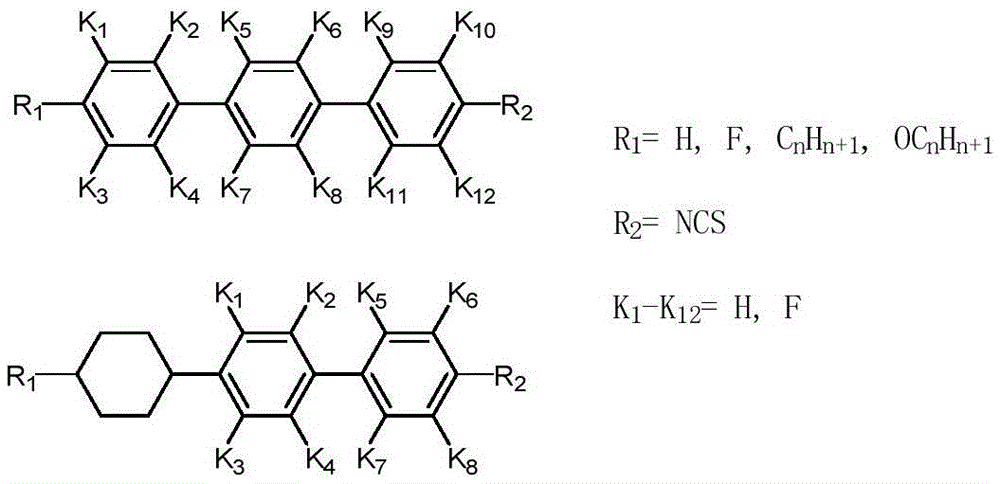
:液晶透镜是一种裸眼3D显示装置中常用的分光器件,其将液晶材料夹在两层电极基板中间,通过电场梯度作用来实现透镜的效果。液晶透镜中用到的高双折射率液晶材料一般为一种小分子化合物的混合物,以下是液晶材料小分子的常见结构,如图1所示,两个带侧链(R1)或端基(R2)的环结构(A1,A2)由连接基团(X)连接起来。图2所示的一类常用异硫氰基类高双折射率液晶材料分子单晶结构。由于异硫氰基类液晶材料的化学结构对温度和紫外光等相对稳定,所以该液晶材料的性能表现良好,由此应用非常广泛,也是现阶段研究最多的一类液晶材料。对于异硫氰基类高双折射率液晶材料的单晶结构,都可以用铃木反应(suzukireaction)获得,如图3中反应式所示。经过suzuki反应得到的中间产物与CSCl2反应,得到最终的高双折射率液晶材料单晶结构。经过suzuki反应后,催化剂钯不易去除,反应底物芳基硼酸在suzuki反应条件下会产生自身偶合的副产物。如果将带有这些杂质的中间产物直接用于合成异硫腈基(NCS)类液晶单体,则产生的副产物用柱层析色谱法很难将它们分离并且产物会发黄,导致最终产物纯化困难并影响纯度,由此对由该液晶材料制作的器件的性能有很大的影响。因此,在合成最终反应产物NCS类液晶单体步骤前,必须要得到高纯度的带有胺基的中间产物。目前,去除钯催化剂和芳基硼酸自身耦合副产物的方法为:将suzuki反应后的产物通过柱层析法,将杂质与中间产物得以分离。但是suzuki反应体系所产生的芳基硼酸自身偶合副产物和容易残留的金属钯会加大分离的难度,所需花费的时间长,使用的溶剂较多。这对于工业化大量生产是十分不利的。因此需提供一个简便有效的分离方法来实现对产物的提纯。技术实现要素:本发明要解决的技术问题是提供一种液晶材料中间体以及该液晶材料的制备方法,该方法操作简单、节省溶剂,且可以得到纯度较高的液晶材料中间体,从而便于在工业化的大量生产中应用。本发明公开了一种液晶材料中间体的制备方法,包括以下步骤:(A)在钯催化剂作用下,将式II所示化合物、式III所示化合物和碱于醇和水的混合物中,加热发生铃木反应,其中,所述醇为C3~C8一元醇或C3~C8二元醇;(B)在上述反应完成后,保持80~90℃静置分层,分离有机相,除去有机相中残留的碱后,在室温下静置,析出式I所示的液晶材料中间体;其中,R1为具有1-7个碳原子的烷基、具有1-7个碳原子的不饱和烃基或具有1-7个碳原子的烷氧基;L1、L2、L3、L4、L5、L6、L7、L8分别为H或F;m为0、1或2;X为-Cl、-Br或-I,为或优选的,所述步骤(A)中,所述醇为C3~C5一元醇或C3~C5二元醇。优选的,所述步骤(A)中,所述醇和水的体积比为2~5:1。优选的,所述步骤(A)具体为:将式III所示化合物、碱及催化剂先溶于所述醇和水的混合物中,然后将式II所示化合物分次加入,加热发生铃木反应。优选的,所述步骤(A)具体为:将式III所示化合物、碱及催化剂先溶于所述醇和水的混合物中,反应1小时得到反应液,然后将式II所示化合物溶于所述醇中,分次加入所述反应液,加热发生铃木反应。优选的,所述步骤(B)中去除有机相中残留的碱的方法具体为:将有机相在80~90℃搅拌析晶,直至不再有晶体析出后,过滤,取滤液在室温下静置,析出式I所示的液晶材料中间体。优选的,所述步骤(B)中去除有机相中残留的碱的方法具体为:萃取所述有机相,水洗除掉所述有机相中残留的碱,旋干后加入所述醇加热溶解,在室温下静置,析出式I所示的液晶材料中间体。优选的,所述步骤(B)中,所述静置分层的时间为1~3小时。优选的,所述步骤(A)中,所述钯催化剂为Pd(Pph3)4、Pb(dppf)Cl2或Pd(Pph3)2Cl2。优选的,所述步骤(A)中,所述碱为Cs2CO3、K2CO3、Na2CO3、Li2CO3、K3PO4或Ba(OH)2。优选的,所述步骤(A)中,所述反应在惰性气体中进行。优选的,所述步骤(A)中,所述反应的时间为10~15小时。本发明还公开了一种液晶材料的制备方法,将上是技术方案所述方法制备的液晶中间体与CSCl2或CS2反应,得到异硫氰基类液晶材料。与现有技术相比,本发明的液晶材料中间体的制备方法具有如下有益效果:本发明采用C3~C8一元醇或二元醇和水的混合物做为溶剂,在铃木反应后,醇与水发生分层,钯催化剂留在水中或者留在醇与水之间的部分,因此直接取上层的有机相即可简单快速的与钯催化剂分离,实现钯催化剂的去除,无需再额外添加溶剂,节省了溶剂。提取的有机相,在高温下将析出多余的盐类,将其过滤后,滤液中仅含有液晶材料中间体,在室温下静置,即可得到纯净的单晶状态的液晶材料中间体。本发明的方法克服了柱层析中费时费力,消耗溶剂多等缺点,达到了操作方便、节省溶剂、获得产物纯净的效果。附图说明图1表示高双折射率液晶材料常见分子结构图;图2表示高双折射率异硫氰基类液晶材料的分子结构图;图3表示异硫氰基类液晶材料的suzuki合成方法。具体实施方式为了进一步理解本发明,下面结合实施例对本发明优选实施方案进行描述,但是应当理解,这些描述只是为进一步说明本发明的特征和优点,而不是对本发明权利要求的限制。本发明公开了一种液晶材料中间体的制备方法,包括以下步骤:(A)在钯催化剂作用下,将式II所示化合物、式III所示化合物和碱于醇和水的混合物中加热发生铃木反应,其中,所述醇为C3~C8一元醇或C3~C8二元醇;(B)在上述反应完成后,保持80~90℃静置分层,分离有机相,除去有机相中残留的碱后,在室温下静置,析出式I所示的液晶材料中间体,其中,R1为具有1-7个碳原子的烷基、具有1-7个碳原子的不饱和烃基或具有1-7个碳原子的烷氧基;L1、L2、L3、L4、L5、L6、L7、L8分别为H或F;m为0、1或2;X为-Cl、-Br或-I,为或在本发明中,所述式I所示的液晶材料中间体为合成异硫氰基类液晶材料时的重要原料,其纯化程度直接影响异硫氰基类液晶材料的纯度。按照本发明,首先在钯催化剂作用下,将式II所示化合物、式III所示化合物和碱于醇和水的混合物中加热发生铃木反应,其中,所述醇为C3~C8一元醇或C3~C8二元醇,优选为C3~C5一元醇或C3~C5二元醇,所述一元醇更优选为异丙醇或丁醇,所述醇和水的体积比优选为2~5:1,更优选为2~3:1。本发明选用的溶剂为醇和水的混合物,有利于铃木反应后钯催化剂和杂质的去除,本发明所使用的醇不限定为C3~C8一元醇或C3~C8二元醇,只要所述醇具备以下条件即可:①对产物溶解性不好,但在高温下可以溶解产物;②反应后静置可以与水分层;③对催化剂不溶。由于式II化合物容易在加热条件下,发生自身偶合,产生副产物,因此为了避免式II所示化合物的自身偶合,优选的:将式III所示化合物、碱及催化剂先溶于所述醇中,然后将式II所示化合物分次加入,加热发生铃木反应。更优选的:将式III所示化合物、碱及催化剂先溶于所述醇和水的混合物中,反应1小时得到反应液,然后将式II所示化合物溶于所述醇中,分次加入所述反应液,加热发生铃木反应。按照该优选的方式,Pd催化剂会先进攻式III所示化合物中卤素,从而形成活泼的中间体,再加入式II所示化合物可以提高反应的产率并减少式II所示化合物自身耦合的副产物。所述反应优选的在惰性气体保护下进行,所述反应的时间优选为10~15小时,所述反应的温度优选为80~90℃。所述钯催化剂优选为Pd(Pph3)4、Pb(dppf)Cl2或Pd(Pph3)2Cl2。所述碱优选为Cs2CO3、K2CO3、Na2CO3、Li2CO3、K3PO4或Ba(OH)2。按照本发明,完成上述铃木反应后,即可对产物进性提纯:将铃木反应的产物保持80~90℃静置分层,分离有机相,去除有机相中残留的碱后,在室温下静置,析出式I所示的液晶材料中间体。中去除有机相中残留的碱的方法优选为:将有机相在80~90℃搅拌析出晶体,直至不再有晶体析出后,过滤,取滤液在室温下静置,析出式I所示的液晶材料中间体。或者还可以优选为:萃取所述有机相,水洗除掉所述有机相中残留的碱,旋干后加入所述醇加热溶解,在室温下静置,析出式I所示的液晶材料中间体。由于铃木反应时采用的为醇和水的混合溶液,因此静置后,两者就会出现分层现象。所述静置分层的时间优选为1~3小时,温度优选为80~90℃,更优选为85℃。经过所述静置分层,如果铃木反应中应用的钯催化剂添加量较少,其会沉积在下层的水中;如果钯催化剂添加量较多,其会沉积在醇和水层之间;而铃木反应中添加的盐或者反应产生的盐大部分会溶解在下层的水相中。静置分层后,分离有机相,所述有机相中即含有式I所示的液晶材料中间体和少量的盐类。所述有机相在80~90℃,优选为85℃搅拌条件下,可以使上述铃木反应中的碱(Cs2CO3、K2CO3、Na2CO3、Li2CO3、K3PO4或Ba(OH)2)析出,而式I所示的液晶材料中间体不析出。最后通过减低温度至室温,析出纯净的单晶结构的式I所示的液晶材料中间体。本发明还公开了一种液晶材料的制备方法,将上述技术方案所述方法制备的液晶中间体与CSCl2或CS2反应,即可得到异硫氰基类液晶材料。为了进一步理解本发明,下面结合实施例对本发明提供的液晶材料中间体及液晶材料的制备方法及柔性显示器件进行详细说明,本发明的保护范围不受以下实施例的限制。实施例1在斜二口烧瓶里放入搅拌子,加入23.14g4-溴苯胺,1.7gPb(dppf)Cl2或Pd(Pph3)2Cl2,15g碳酸钠;搭建回流反应装置,并密封反应系统;将密封系统里的空气置换成氮气,注入700ml异丙醇,350ml水;慢慢加热回流,确保系统在氮气保护下反应。1小时后,将27.4g4-(反式-4-戊基环己基)苯硼酸溶解于异丙醇中形成溶液,分三次加入4-(反式-4-戊基环己基)苯硼酸溶液,每次间隔半小时,继续反应12小时。TLC监控反应过程,反应结束后,静置1小时分层后,将反应上层液倒入布氏漏斗将少量的黑色固体Pd滤掉,滤液加50ml异丙醇继续85℃搅拌,将多余的盐析出来后用布氏漏斗过滤,滤液室温下静置,将析出的单晶产物过滤,即可得到高纯度的单晶产物,滤液可继续重结晶,获得纯度99%以上单晶产物。产率70%。比较例1柱层析色谱法:采用硅胶色谱柱分离的方法,通过TLC点板,反应后共有三个点,产物点在展开剂为石油醚中IF值为0.2处,将350g硅胶湿法装柱,反应后的样品用二氯甲烷和饱和氯化钠水溶液萃取后,有机相干燥,旋干,加硅胶干法拌样上柱,以石油醚为淋洗剂进行色谱分离,得到IF值为0.2处的产物,该过程需要耗时15-20小时,且消耗石油醚20L左右。产率85%。异丙醇结晶法与柱层析色谱法对比:时间周期/mol溶剂消耗量/mol产率其他异丙醇结晶法2-3小时异丙醇约1L80%(纯度99%)柱层析色谱法15-20小时石油醚20L以上85%(纯度97%)需消耗硅胶实施例2在斜二口烧瓶里放入搅拌子,加入22.88g2,6-二氟-4-溴苯胺,1.7gPb(dppf)Cl2或Pd(Pph3)2Cl2,15.9g碳酸钠;搭建回流反应装置,并密封反应系统;将密封系统里的空气置换成氮气,注入700ml异丙醇,350ml水;慢慢加热回流,确保系统在氮气保护下反应。1小时后,将27.4g4-戊基联苯硼酸溶解于异丙醇中形成溶液,分三次加入4-戊基联苯硼酸溶液,每次间隔半小时,继续反应12小时。TLC监控反应过程,反应结束后,静置1小时,将反应上层液倒入布氏漏斗将黑色固体Pd滤掉,滤液加50ml异丙醇继续85℃搅拌,将多余的盐析出来后用布氏漏斗过滤,滤液室温下静置,将析出的单晶产物过滤,即可得到纯的产物单晶,滤液旋掉三分之一溶剂后继续室温结晶,获得纯度99%以上单晶产物。产率70%。比较例2柱层析色谱法:采用硅胶色谱柱分离的方法,通过TLC点板,反应后共有三个点,产物点在展开剂为石油醚:二氯甲烷=50:1中IF值为0.3处,将400g硅胶湿法装柱,反应后的样品用二氯甲烷和饱和氯化钠水溶液萃取后,有机相干燥,旋干,加硅胶干法拌样上柱,以石油醚:二氯甲烷=50:1为淋洗剂进行色谱分离,得到IF值为0.3处的产物,该过程需要耗时15-20小时,且消耗石油醚和二氯甲烷20L左右。产率85%。异丙醇结晶法与柱层析色谱法对比:实施例3在斜二口烧瓶里放入搅拌子,加入20.79g2-氟-4-溴苯胺,1.7gPb(dppf)Cl2或Pd(Pph3)2Cl2,15.9g碳酸钠;搭建回流反应装置,并密封反应系统;将密封系统里的空气置换成氮气,注入700ml丁醇,350ml水;慢慢加热回流,确保系统在氮气保护下反应。1小时后,将16.4g4-丙基苯硼酸溶解于丁醇中形成溶液,分三次加入4-丙基苯硼酸,每次间隔半小时,继续反应12小时。TLC监控反应过程,反应结束后,静置1小时,将反应上层液倒入布氏漏斗将黑色固体Pd滤掉,滤液继续85℃搅拌,将多余的盐析出来后用布氏漏斗过滤,滤液室温下静置,将析出的单晶产物过滤,即可得到纯的产物单晶,滤液旋掉三分之一溶剂后继续室温结晶,获得纯度96%以上单晶产物。产率80%。比较例3柱层析色谱法:采用硅胶色谱柱分离的方法,通过TLC点板,反应后共有三个点,产物点在展开剂为石油醚:二氯甲烷=30:1中IF值为0.3处,将400g硅胶湿法装柱,反应后的样品用二氯甲烷和饱和氯化钠水溶液萃取后,有机相干燥,旋干,加硅胶干法拌样上柱,以石油醚:二氯甲烷=30:1为淋洗剂进行色谱分离,得到IF值为0.3处的产物,该过程需要耗时15-20小时,且消耗石油醚和二氯甲烷18L左右。产率85%。异丙醇结晶法与柱层析色谱法对比:实施例4在斜二口烧瓶里放入搅拌子,加入22.88g2,4-二氟-4-溴苯胺,1.7gPb(dppf)Cl2或Pd(Pph3)2Cl2,15.9g碳酸钠;搭建回流反应装置,并密封反应系统;将密封系统里的空气置换成氮气,注入800ml丁醇,350ml水;慢慢加热回流,确保系统在氮气保护下反应。1小时后,将24g4-丙基苯硼酸分三次加入,每次间隔半小时,继续反应12小时。TLC监控反应过程,反应结束后,静置1小时,分层后,萃取有机相,用水析出掉所述有机相中残留的碳酸钠,旋干后加入150ml丁醇,加热溶解,在室温下静置,析出结晶,获得纯度99%以上单晶产物。产率88%。比较例4柱层析色谱法:采用硅胶色谱柱分离的方法,通过TLC点板,反应后共有三个点,产物点在展开剂为石油醚:二氯甲烷=30:1中IF值为0.2处,将400g硅胶湿法装柱,反应后的样品用二氯甲烷和饱和氯化钠水溶液萃取后,有机相干燥,旋干,加硅胶干法拌样上柱,以石油醚:二氯甲烷=30:1为淋洗剂进行色谱分离,得到IF值为0.2处的产物,该过程需要耗时15-20小时,且消耗石油醚和二氯甲烷25L左右。产率88%。异丙醇结晶法与柱层析色谱法对比:以上实施例的说明只是用于帮助理解本发明的方法及其核心思想。应当指出,对于本
技术领域:
的普通技术人员来说,在不脱离本发明原理的前提下,还可以对本发明进行若干改进和修饰,这些改进和修饰也落入本发明权利要求的保护范围内。对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1