受柠檬酸钠诱导的启动子的制作方法
本发明涉及受柠檬酸钠诱导的启动子。
背景技术:
在利用基因工程菌株表达目标蛋白时,目标都是希望以尽可能高的效率来产出多的目的蛋白,许多因素可以影响蛋白的产量,例如启动子活性、翻译效率、蛋白糖基化、目标基因的拷贝数以及菌株自身蛋白酶的降解能力,其中以启动子转录水平的高低影响最为关键。
启动子是与rna聚合酶结合的一段dna序列,参与特定基因转录及调控,包含核心启动子区域和调控区域,核心启动子区域产生基础水平的转录,调控区域能够对不同的环境条件做出应答,对基因的转录水平做出相应的调节。
曲霉中已有许多强启动活性的启动子被鉴定并分离出来,包括组成型启动子和诱导型启动子两类,组成型启动子是一类无需在培养基中添加任何诱导物便可自发调控结构基因进行表达的启动子,曲霉蛋白表达中最常见的组成型启动子有构巢曲霉甘油醛-3-磷酸脱氢酶gpda启动子,泡盛曲霉谷氨酸脱氢酶gdha启动子等〔雷达,许杨等,曲霉属蛋白表达中高效启动子的应用研究[j],食品工业科技,2013,34(13):342-345〕,但组成型启动子调控下菌体生长和目的蛋白的表达同时进行,所以往往伴随着效率不高的问题。诱导型启动子则是另外一种可以受诱导物调控表达的启动子,黑曲霉的葡糖淀粉酶基因glaa启动子和米曲霉的淀粉酶taka启动子是目前曲霉表达系统中使用率最高的诱导型启动子,但这些启动子通常需要大量添加淀粉、糊精或麦芽糖等碳源来诱导表达,使得培养基粘度大大提高,影响溶氧和后处理工艺。
柠檬酸钠,又称枸橼酸钠,是一种有机化合物,外观为白色到无色晶体。根据《食品安全国家标准、食品添加剂使用标准》(gb2760-2011)规定:可在各类食品中按生产需要适量使用的食品添加剂。
柠檬酸钠主要由淀粉类物质经发酵生成柠檬酸,再跟碱类物质中和而产生,具有以下优良性能:(1)安全无毒性能。由于制备柠檬酸钠的原料基本来源于粮食,因而绝对安全可靠,对人类健康不会产生危害。联合国粮农与世界卫生组织对其每日摄入量不作任何限制,可认为该品属于无毒品。(2)具有生物降解性。柠檬酸钠经自然界大量的水稀释后,部分变成柠檬酸,两者共存于同一体系中。柠檬酸在水中经氧、热、光、细菌以及微生物的作用,很容易发生生物降解。(3)具有金属离子络合能力。柠檬酸钠对ca2+、mg2+等金属离子具有良好的络合能力。(4)极好的溶解性能,并且溶解性随水温升高而增加。(5)具有良好的ph调节及缓冲性能。柠檬酸钠是一种弱酸强碱盐,与柠檬酸配伍可组成较强的ph缓冲剂,因此在某些不适宜ph大范围变化的场合有其重要用处。另外,柠檬酸钠还具有优良的缓凝性能及稳定性能。
柠檬酸钠具有上述多种优良的性能,使得其具有十分广泛的用途。柠檬酸钠无毒性、具有ph调节性能及良好的稳定性,因此可用于食品工业。柠檬酸钠用作食品添加剂,需求量最大,主要用作调味剂、缓冲剂、乳化剂、膨胀剂、稳定剂和防腐剂等;另外,柠檬酸钠同柠檬酸配伍,用作各种果酱、果冻、果汁、饮料、冷饮、奶制品和糕点等的胶凝剂、营养增补剂及风味剂〔张英,周长民,柠檬酸钠的特性与应用[j],辽宁化工,2007.5,36(5):350-352〕。
技术实现要素:
本发明提供一种核酸分子,含有选自以下的多核苷酸序列:
(1)seqidno:1-4中任一所示的多核苷酸序列;和
(2)(1)所述多核苷酸序列的互补序列。
本发明还提供一种核酸构建体,所述核酸构建体含有本发明所述的核酸分子作为该核酸构建体的启动子序列。
在某些实施方案中,所述核酸构建体还含有多克隆位点或至少一个酶切位点,翻译起始用的核糖体结合位点,3’转录终止子,3’多聚核苷酸化信号,转运和靶向核酸序列,抗性选择标记,增强子或操作子中的任一个、任意数个或全部。
在某些实施方案中,所述核酸构建体是表达载体。
在某些实施方案中,所述核酸构建体还含有感兴趣的基因,与所述启动子序列操作性连接。
在某些实施方案中,所述感兴趣的基因选自脂肪酶基因、蛋白酶基因、酯酶基因、淀粉酶基因、果胶酶基因、植酸酶基因、糖化酶基因、漆酶基因、单宁酶基因、半乳糖苷酶基因、葡聚糖酶基因、阿拉伯呋喃糖苷酶基因、氨基肽酶基因、多聚半乳糖醛酸酶基因、过氧化氢酶基因、菊糖酶基因、木聚糖酶基因、凝乳酶基因、葡糖氧化酶基因、普鲁兰酶基因、乳糖酶基因、天门冬酰胺酶基因、脱氨酶基因、磷脂酶基因和纤维素酶基因。
在某些实施方案中,所述感兴趣的基因为来自丝状真菌,如黑曲霉、米曲霉、构巢曲霉、里氏木霉、土曲霉、米黑根毛霉等的脂肪酶基因。
本发明还提供遗传工程化的宿主细胞,所述宿主细胞含有本发明的核酸构建体或表达载体。
本发明还提供一种遗传工程化的丝状真菌,该丝状真菌转化有本发明的核酸构建体表达载体。
在某些实施方案中,所述丝状真菌选自:黑曲霉、米曲霉、构巢曲霉、里氏木霉、土曲霉和米黑根毛霉。
本发明还提供一种减少丝状真菌发酵中淀粉、糊精或麦芽糖等碳源的使用量的方法,所述方法包括使用本发明的丝状真菌进行发酵。
本发明还提供一种丝状真菌发酵方法,所述方法包括在发酵培养基中添加柠檬酸钠作为诱导剂发酵本发明的丝状真菌。
本发明还包括本发明核酸分子和核酸构建体在减少丝状真菌发酵中碳源,如淀粉、糊精或麦芽糖的使用量中的用途。
本发明也包括柠檬酸钠在诱导目的蛋白表达中的用途,尤其是在权利要求1所述的核酸分子调控的目的蛋白的表达中作为诱导物的用途
附图说明
图1显示启动子验证表达载体示意图。
图2显示脂肪酶活力测定方法流程示意图。
图3显示启动子活力检测柱形图。
图4显示启动子诱导效果柱形图。
具体实施方式
本发明从黑曲霉(aspergillusniger)中分离得到可作为启动子使用的核酸序列。使用柠檬酸钠可诱导本发明的启动子表达。
如本文所用,“启动子”指一种核酸序列,其通常存在于目的基因编码序列的上游(5’端),能够引导目的基因转录为mrna。一般地,启动子区提供rna聚合酶和正确起始转录所必需的其它因子的识别位点。如本文所用,“分离的”是指物质从其原始环境中分离出来(如果是天然的物质,原始环境即是天然环境)。如活体细胞内的天然状态下的多聚核苷酸和多肽是没有分离纯化的,但同样的多聚核苷酸或多肽如从天然状态中同存在的其他物质中分开,则为分离纯化的。
本发明的核酸分子的多核苷酸序列如seqidno:1-4中任一条所示。此外,本发明还包括上述核酸分子的一些具有相同功能的变异体。包括:
在严格条件下能与seqidno:1、seqidno:2、seqidno:3或seqidno:4所示的多核苷酸序列杂交,优选在严格条件下杂交,且能指导目的基因表达的核酸分子;
与seqidno:1、seqidno:2、seqidno:3或seqidno:4所示的多核苷酸序列有80%以上,优选至少85%以上,优选至少90%以上,优选至少95%以上,优选至少96%以上,优选至少97%以上,优选至少98%以上,或优选至少99%以上的同一性,且能指导目的基因功能的核酸分子;和
与seqidno:1、seqidno:2、seqidno:3或seqidno:4所示的多核苷酸序列互补(优选完全互补)的核酸分子。
在本发明中,“严格条件”是指:(1)在较低离子强度和较高温度下的杂交和洗脱,如0.2×ssc,0.1%sds,60℃;或(2)杂交时加有变性剂,如50%(v/v)甲酰胺,0.1%小牛血清/0.1%ficoll,42℃等;或(3)仅在两条序列之间的同一性至少在90%以上,更好是95%以上时才发生杂交。并且,可杂交的核酸也具有指导目的基因高水平表达的功能。
可采用常规的技术确定序列之间的序列同一性,例如通过ncbi网站上提供的序列比对软件(nucleotideblast)等确定序列之间的同一性。
优选的是,所述变异体的突变发生在启动子的调控区域,例如可通过插入或删除调控区域,或对调控区域进行随机或定点突变等来获得。换言之,本发明也包括在seqidno:1、seqidno:2、seqidno:3或seqidno:4所示的多核苷酸序列的基础上进行一个、数个或数十个(例如100个以内,或者80个以内,或60个以内,或50个以内,或40个以内,或30个以内,或20个以内,或15个以内,或10个以内,或5个以内)的碱基删除或突变而获得的保留了启动子指导目的基因表达的功能的seqidno:1、seqidno:2、seqidno:3或seqidno:4的变异体。
本发明也包括seqidno:1-4的长例如15~50个碱基的片段,更优选长15~30个碱基的片段。
本发明的启动子可以被操作性连接到目的基因上。如本文所用,“操作性连接”指两个或多个核酸区域或核酸序列的功能性的空间排列。例如:启动子被置于相对于目的基因核酸序列的特定位置,使得核酸序列的转录受到该启动子区域的引导,从而,启动子被“操作性连接”到该核酸序列上。
该目的基因相对于启动子而言可以是外源(异源)的。适用于本发明的目的基因通常可以是任何核酸序列(如一种结构性核酸序列),优选是编码具有特定功能的蛋白,例如某些具有重要特性或功能的蛋白。
本发明感兴趣的目的基因包括但不限于本领域周知的各种功能蛋白的编码基因,尤其是本领域常在微生物(例如丝状真菌)中表达、生产的蛋白的编码基因,包括各种酶的编码基因。因此,感兴趣的目的基因包括但不限于脂肪酶基因、蛋白酶基因、酯酶基因、淀粉酶基因、果胶酶基因、植酸酶基因、糖化酶基因、漆酶基因、单宁酶基因、半乳糖苷酶基因、葡聚糖酶基因、阿拉伯呋喃糖苷酶基因、氨基肽酶基因、多聚半乳糖醛酸酶基因、过氧化氢酶基因、菊糖酶基因、木聚糖酶基因、凝乳酶基因、葡糖氧化酶基因、普鲁兰酶基因、乳糖酶基因、天门冬酰胺酶基因、脱氨酶基因、磷脂酶基因和纤维素酶基因等。
这些感兴趣的基因可以来自不同的物种来源,包括真核生物和原核生物。在某些实施方案中,所述感兴趣的基因优选为脂肪酶基因和磷脂酶基因。例如,所述感兴趣的基因是来自丝状真菌,如黑曲霉、米曲霉、构巢曲霉、里氏木霉、土曲霉、米黑根毛霉等的脂肪酶基因,如来自米黑根毛霉的脂肪酶基因。
因此,本发明也提供一种核酸构建体,该核酸构建体含有本发明的启动子。优选的是,该核酸构建体还含有:在所述启动子的下游包含多克隆位点或至少一个酶切位点,翻译起始用的核糖体结合位点,3’转录终止子,3’多聚核苷酸化信号,其它非翻译核酸序列,转运和靶向核酸序列,抗性选择标记,增强子或操作子中的任一个、任意数个或全部。
当需要表达目的基因时,将目的基因连接入适合的多克隆位点或酶切位点内,从而将目的基因与启动子可操作地连接。因此,在一个具体实施例中,本发明的核酸构建体还包括感兴趣的目的基因,本发明的启动子能指导所述感兴趣的目的基因在宿主细胞中表达。
合适的转录终止子序列是由宿主细胞识别以终止转录的序列。终止子序列与感兴趣的目的基因的3′末端操作性连接。在选择的宿主细胞中有功能的任何终止子都可用于本发明。
例如,用于细菌宿主的优选终止子可以是来自t7噬菌体的终止子。
用于丝状真菌宿主细胞的优选终止子获得自米曲霉taka淀粉酶、黑曲霉葡萄糖淀粉酶、构巢曲霉邻氨基苯甲酸合酶、黑曲霉α-葡萄糖苷酶的基因。
用于酵母宿主细胞的优选终止子获得自酿酒酵母烯醇酶、酿酒酵母细胞色素c、酿酒酵母甘油醛-3-磷酸脱氢酶、巴斯德毕赤酵母醇氧化酶基因等。
核酸构建体还可包括合适的前导序列,前导序列与感兴趣的目的基因的5′末端操作性连接。在选择的宿主细胞中有功能的任何终止子都可用于本发明。
核酸构建体还可包括信号肽编码区,该编码区编码的氨基酸序列与感兴趣的目的基因编码的多肽的氨基酸末端连接并指导该编码的多肽进入细胞分泌途径。感兴趣的目的基因的5′端可固有地包含信号肽编码区,或者也可使用外源的信号肽编码区。外来的信号肽编码区可简单地替换天然的信号肽编码区以增强多肽的分泌。指导表达的多肽进入选择的宿主细胞的分泌途径的任何信号肽编码区,即分泌进入培养基,都可用于本发明。
本发明的核酸构建体优选为表达载体。术语“表达载体”指本领域熟知的细菌质粒、噬菌体、酵母质粒、病毒或其他载体。质粒可以是线性或闭合的环形质粒。总之,只要其能够在宿主体内复制和稳定,任何质粒和载体都是可以被采用的。载体的选择一般取决于载体与其中被导入该载体的宿主细胞的相容性。
表达载体一般包括(从5’到3’方向):引导目的基因转录的启动子和目的基因。如果需要,所述的重组载体还可以包括:在所述启动子的下游包含多克隆位点或至少一个酶切位点,翻译起始用的核糖体结合位点,3’转录终止子,3’多聚核苷酸化信号,其它非翻译核酸序列,转运和靶向核酸序列,抗性选择标记,增强子或操作子。目的基因连接入所述适合的多克隆位点或酶切位点内,从而将目的基因与启动子可操作地连接。
本领域常用的各种翻译起始用的核糖体结合位点、3’多聚核苷酸化信号,其它非翻译核酸序列,转运和靶向核酸序列,抗性选择标记,增强子或操作子都可用于本发明。
载体可以是自主复制的载体,即作为染色体外实体存在,其复制不依赖于染色体复制的载体,例如质粒、染色体外元件、微型染色体或人工染色体。载体可包含用于保证自我复制的任何方式。备选地,载体可以是当被导入宿主细胞时,整合到基因组中并且与其已经被整合进入的染色体一起复制的载体。此外,可使用一起包含将被导入宿主细胞基因组的总dna的单个载体或质粒或两个或多个载体或质粒,或转座子。
本发明的载体优选包含一个或多个容许容易选择转化、转染、转导等细胞的可选择标记。可选择的标记是基因,其产物提供对抗生素或病毒的抗性、对重金属的抗性、原养型至营养缺陷型等。
本发明的载体优选包含容许该载体整合进入宿主细胞基因组或该载体在细胞中独立于基因组自主个复制的元件。
表达载体可含有一个以上拷贝的感兴趣的目的基因,以增加该基因产物的产量。目的基因拷贝数的增加可通过将至少一个附加拷贝的序列整合进入宿主细胞基因组或通过包括可扩增的选择标记基因和该目的基因来获得,其中包含扩增拷贝的选择标记基因并且由此包含附加拷贝目的基因的细胞可通过在存在适当的选择剂时培养该细胞来筛选。
用于制备重组表达载体的方法是本领域技术人员所熟知的。这些方法包括体外重组dna技术、dna合成技术、体内重组技术等。
作为表达载体的一个例子,本发明使用ncbi序列号为fj868795.1的表达载体构建了含有本发明启动子序列的表达载体。因此,应理解的是,除fj868795.1所含的米黑根毛霉脂肪酶(rml)基因外,本发明可使用其它感兴趣的基因替换该基因,从而以该表达载体为骨架,骨架含有本发明启动子序列和不同感兴趣基因的表达载体。
本发明也涉及经本发明表达载体转化的重组宿主细胞。可采用本领域周知的方法进行转化,例如接种市售的转染试剂进行转化。
宿主细胞可以是单细胞微生物或非单细胞微生物。单细胞微生物例如革兰氏阳性细菌,包括但不限于芽孢杆菌细胞,例如,嗜碱芽孢杆菌、解淀粉芽孢杆菌、短芽孢杆菌、巨大芽孢杆菌、枯草芽孢杆菌、地衣芽孢杆菌、凝结芽孢杆菌、嗜热脂肪芽孢杆菌和苏云金芽孢杆菌等;或链霉菌细胞,例如钱青紫链霉菌;或革兰氏阴性细菌,例如大肠杆菌和假单胞菌属。在优选的方面,细菌宿主是枯草芽孢杆菌、大肠杆菌、地衣芽孢杆菌、嗜热脂肪芽孢杆菌和大肠杆菌细胞。
宿主细胞也可以是真核生物的细胞,例如来自哺乳动物、昆虫、植物、酵母或真菌的细胞。
在优选的方面,本发明提供遗传工程化的丝状真菌,如黑曲霉、米曲霉、构巢曲霉、里氏木霉、土曲霉、米黑根毛霉等。所述丝状真菌经转化含有本发明的表达载体。转化的方法为本领域常规的转化方法。作为一个示范性的例子,本申请的实施例5提供了将本发明表达载体转化到丝状真菌,具体是黑曲霉中的具体方法。但应理解,本发明并不仅限于该方法。
本发明还提供一种减少丝状真菌发酵中碳源,如淀粉、糊精或麦芽糖等碳源的使用量的方法,所述方法包括使用本发明的丝状真菌进行发酵,并添加柠檬酸钠作为诱导剂。
本发明还提供一种丝状真菌发酵方法,所述方法包括在发酵培养基中添加柠檬酸钠作为诱导剂发酵本发明的丝状真菌。
柠檬酸钠的添加量可根据实际的发酵情况(例如用于发酵的真菌、发酵的培养基等)确定,通常占所用培养基重量的1~10%,如1~5%。
本发明因此也包括柠檬酸钠在诱导目的蛋白表达中的用途,包括作为诱导型启动子的诱导物中的用途。所述诱导型启动子包括但不限于本文所述的核酸分子,更优选为seqidno:1-4任一所示的核酸分子。因此,本发明尤其包括柠檬酸钠在本发明核酸分子(如seqidno:1-4中任一所示的序列)调控的目的蛋白的表达中的用途。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明中。文中所述的较佳实施方法与材料仅作示范之用。
1、实验菌株和质粒
菌株名称:黑曲霉as3.975(cgmcc:3.795);
质粒:p3sr2,含乙酰胺酶(amds)基因(bccm/lmbp:accessionnumber:2363),psp72。
2、培养基和溶液
菌丝培养基(1l):胰蛋白胨20g,酵母抽提物10g,20%葡萄糖100ml,115℃,高压灭菌15min。
再生培养基:0.1%磷酸氢二钾、0.05%氯化钾、0.05%硫酸镁、0.001%硫酸亚铁、1m蔗糖、1.5%琼脂粉,115℃,高压灭菌15min。待冷却后加入终浓度15mm的乙酰胺和20mm的cscl。
pda-rhodamineb筛选培养基:
pda培养基:马铃薯(去皮)200g,葡萄糖20g,琼脂20g,水1000ml,ph值自然。
橄榄油乳化液:以1:3体积比加入橄榄油与4%的聚乙烯醇(pva)溶液并混合,用高速匀浆机8000rpm匀浆15min,间歇5min后再搅拌3min。
pda培养基和橄榄油乳化液分别115℃,高压灭菌15min;待冷却至60℃左右,按9:1加入pda培养基和橄榄油乳化液,再加入1/100体积的过滤灭菌的0.1%rhodamineb溶液,混匀后立即倒平板。
黑曲霉发酵培养基:2%葡萄糖,2%麦芽糖,1.5%硫酸铵,4%胰蛋白酶消化的大豆培养液,0.1%磷酸二氢钠,0.1%硫酸镁,0.07%tween80,微量元素,115℃,高压灭菌15min(三角瓶为挡板三角瓶,装液量50ml/250ml)。
孢子洗液:可用生理盐水,0.9%nacl,0.05%吐温80。
渗透压稳定剂:0.6mmgso4,10mmnah2po4,ph=5.8。
酶解液:用渗透压稳定剂配制1%裂解酶,1%纤维素酶,0.1%蜗牛酶的酶解液,用0.22μm的微孔滤膜过滤除菌。
山梨醇溶液:1.0m山梨醇,100mmtris-hcl,ph=7.0。
stc:1.0m山梨醇,50mmcacl2,50mmtris-hcl,ph=7.5。
ptc:40%peg4000,50mmcacl2,50mmtris-hcl,ph=7.5。
实施例1:柠檬酸钠的诱导表达
在黑曲霉发酵培养基中添加2%的柠檬酸钠作为诱导培养基,而未添加柠檬酸钠的为未诱导培养基。
用孢子洗液洗脱新鲜的黑曲霉3.795的孢子,通过mircloth过滤制备成孢子悬液,并调整至1×107个/ml。
分别接种1ml黑曲霉孢子悬液至诱导和未诱导培养基中,28℃,200rpm,发酵3天,收集菌丝,得到诱导组样品和未诱导组样品。
实施例2:rna的提取
按照rneasyminikitforpurificationoftotalanimalcells,animaltissues,bacteria,andyeast,andforrnacleanup(qiagen公司)产品说明书实验步骤提取诱导组样品与未诱导组样品的rna。
实施例3:转录组测序及分析
转录组测序采用美国illumina公司的第二代测序技术hiseq2000平台,操作流程参考illumina官方指导手册。提取的rna通过安捷伦2100质检合格后,使用随机引物pd(n)6(含有6个碱基的随机序列)对转录组进行扩增,按照truseqrnakit使用说明构建测序300-500bp的测序文库(http://www.illumina.com/products/truseq_rna_sample_prep_kit_v2.html)。使用黑曲霉参考菌株(cbs513.88)作为参考基因组序列(ncbi索取号:am270980–am270998),并利用比对和表达差异分析软件(tophat2和cufflinks)对于目的菌株的表达差异进行分析。根据差异表达分析结果,选择表达差异倍数(foldchange)大于2倍,pvalue<0.05的基因作为候选基因进行启动子分析。下表1显示通过分析获得的4个候选基因。
表1
实施例4:启动子表达载体的构建
通过pcr技术分别扩增获得4个候选基因的启动子序列,扩增模板为黑曲霉as3.975基因组dna。
引物如下:
p881-f:5'ccgctcgagatcagcaatgccactcgatca3'(seqidno:5)
p881-r:5'cccaagcttgctgaaatgattgaatggctg3'(seqidno:6)
p2870-f:5'ccgctcgagatgcgcccatcaaccgtatgt3'(seqidno:7)
p2870-r:5'cccaagctttagagatagcagggaagcggt3'(seqidno:8)
p4032-f:5'ccgctcgaggatcatcctcctccgtctcga3'(seqidno:9)
p4032-r:5'cccaagcttgatggcagtttctcggctctc3'(seqidno:10)
p4772-f:5'ccgctcgagtactttgatggcggtgcagtc3'(seqidno:11)
p4772-r:5'cccaagcttattgacagagagtgcaggggc3'(seqidno:12)
pcr扩增结果获得的启动子序列长度都约1.6kb大小,其序列分别如seqidno:1-4所示。扩增时添加xhoi和hindiii酶切位点以便后续构建使用。
用xhoi和hindiii酶切pcr扩增获得的启动子序列并连接到同样用xhoi和hindiii酶切的含有米黑根毛霉(rhizormucormiehei)脂肪酶(rml)基因和米曲霉糖化酶终止子的表达载体(ncbi序列号:fj868795.1)上。启动子表达载体的构建如图1所示。
另外,以相同的构建将淀粉酶启动子(ptaka)和糖化酶启动子(pglaa)插入表达框作为对照。
实施例5:黑曲霉的转化
用孢子洗液洗脱新鲜的黑曲霉as3.795孢子,通过mircloth过滤制备成孢子悬液,并调整至1×107个/ml。接种1ml孢子悬液至菌丝培养基中,28℃,180rpm,培养20-24小时,用灭菌mircloth过滤收集长出的菌丝。
收集的菌丝体用灭菌的渗透压稳定剂冲洗三次,压干。菌丝转移至100ml三角瓶中,每0.8g菌丝体重悬在20ml的酶解液中,30℃,60rpm,60-90min。酶解好的原生质体混合液用mircloth过滤,收集滤液,4℃,1000g离心10min,用5ml预冷1.0mol/l山梨醇溶液重悬原生质体沉淀,800g,4℃离心10min,弃上清。再用预冷的stc溶液将原生质体调整至适宜浓度(1×107个/ml),冰浴待用。
向200μl原生质体悬液中,加入20μldna,启动子表达载体和筛选质粒p3sr2各10μl(1μg/μl),再添加50μlptc(peg,tris-hcl,cacl2溶液)溶液,混匀后冰浴30min。加入0.2mlptc溶液,混匀后再加入0.8mlptc溶液,混匀,室温保持30min。
上述混合液中加入200μl再生培养基(上层,0.6%琼脂糖)中,铺于再生培养基(1.5%琼脂糖)上,28℃,培养10天,待菌落长出,统计转化子。
将在平板上长出的菌落,转接至pda-rhodamineb筛选培养基上,28℃,培养3天最终选取在紫外下具有荧光的转化子菌落。
将转化子在pda-rhodamineb筛选培养基上再进行单孢分离纯化后,选取荧光最强的单菌落进行后续发酵试验。
实施例6:启动子活性发酵检测
用孢子洗液洗脱各启动子的转化子和对照转化子的新鲜孢子,通过mircloth过滤制备成孢子悬液,并调整至1×107个/ml。接种1ml孢子悬液到50ml浓度为2%的黑曲霉柠檬酸钠发酵培养基中,对照用淀粉酶启动子和糖化酶启动子培养基中添加2%的淀粉(每个浓度做三个平行样),28℃,200rpm发酵培养96h后取样。
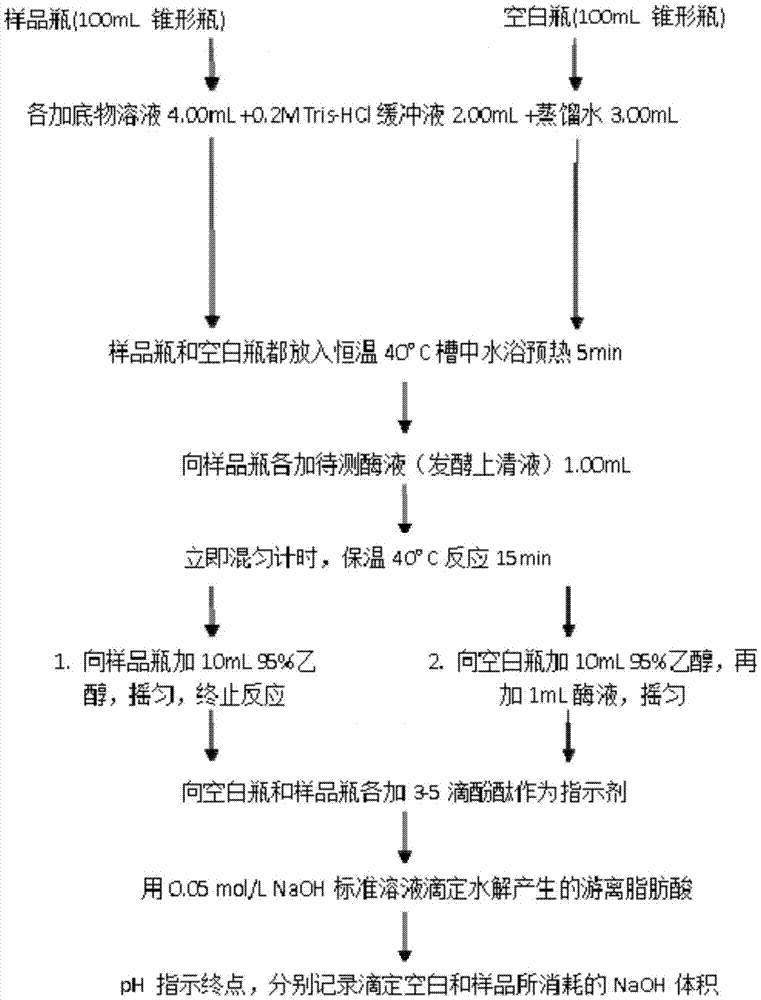
各取1ml黑曲霉摇瓶发酵液到离心管中,12000rpm离心1min;取上清,根据酶活取0.5-5u脂肪酶水解活力的发酵液到总体积1ml的无菌水,作为酸碱滴定法检测酶活的样品。
脂肪酶水解活力酸碱滴定法测定:方法参考qb/t1803-1993工业酶制剂通用试验方法,稍作修改,将缓冲液换成0.2mmol/l的tris-hcl,ph8.0。
橄榄油底物溶液:量取4%pva溶液150ml,加橄榄油50ml,用高速匀浆机8000rpm匀浆15min,制备底物溶液(此溶液需现配现用)。
具体步骤如图2所示。
脂肪酶酶活力单位定义为:每分钟催化底物释放出1μmol脂肪酸的酶量为1个脂肪酶活力单位(u)。酶活计算公式:
样品的酶活(u/ml)=〔(v-v0)/t×n〕×m
式中:v:滴定样液所消耗的naoh溶液体积(ml);v0:滴定空白样所消耗的naoh溶液体积(ml);t:反应时间(min);n:酶液体积(ml);m:滴定用的naoh溶液的浓度(mmol/l)。
在此不同的启动子的活力表现是通过脂肪酶的酶活来体现的,具体酶活见图3。
由图可以得出p2870的启动子活力比较理想,与曲霉中常用的淀粉酶启动子和糖化酶启动子相比,启动子活力能提高大约1倍。
实施例7:启动子诱导效果检测
与实施例6中操作相同,将各启动子的转化子接种到有诱导物和没有诱导物的发酵培养基中,进行发酵培养。p881、p2870、p4032、p4772的诱导物为2%的柠檬酸钠,ptaka和pglaa的诱导物为2%的淀粉。
发酵后进行脂肪酶活力检测,具体方法参考实施例6,启动子诱导效果的结果见图4。
由图可以得出,p881、p2870、p4032、p4772均明显能够受柠檬酸钠的诱导,且p881和p2870与对照相比,诱导效果更加严谨。
- 还没有人留言评论。精彩留言会获得点赞!
- 大豆低温诱导型启动子、包含该启动子的重组表达载体及应用的制作方法与工艺
- 受植物激素调控的启动子及其应用的制作方法与工艺
- 一种受盐诱导的特异性诱导型启动子DNA序列及应用的制作方法与工艺
- 受维甲酸调控的具有浓度依赖效应的重组启动子及其使用的制作方法与工艺
- 一种磷酸盐诱导启动子降低黑曲霉β‑葡萄糖苷酶表达酶活的方法与流程
- 一种疫霉诱导性人工合成启动子PMP1及其重组表达载体和应用的制作方法与工艺
- 一种植物盐/干旱诱导型启动子soyEQ及其应用的制作方法与工艺
- 新型的L-苏氨酸-诱导型启动子及其应用的制作方法与工艺
- 一种植物茉莉酸甲酯诱导表达启动子PosMeJ1及其应用的制作方法与工艺
- 一个逆境诱导启动子SlWRKY31P的克隆及应用的制造方法与工艺