作为治疗剂的人雄激素受体DNA‑结合结构域(DBD)化合物及其使用方法与流程
本申请要求于2014年02月14日提交的美国临时专利申请号61/940,275和2014年07月11日提交的美国临时专利申请号62/023,773的权益,这两篇文献的发明名称均为“作为治疗剂的人雄激素受体二聚体结合结构域(DBD)化合物及其使用方法”。
技术领域
本发明涉及治疗化合物和组合物及其用于治疗各种适应证、包括各种癌症的方法。特别地,本发明涉及用于治疗癌症、例如前列腺癌的疗法和方法。
背景
已知雄激素通过雄激素受体(AR)介导它们的作用。雄激素在广泛的发育和生理响应中发挥作用并且参与男性性分化、维持精子发生和男性促性腺激素调节(R.K.Ross,G.A.Coetzee,C.L.Pearce,J.K.Reichardt,P.Bretsky,L.N.Kolonel,B.E.Henderson,E.Lander,D.Altshuler&G.Daley,Eur Urol 35,355-361(1999);A.A.Thomson,Reproduction 121,187-195(2001);N.Tanji,K.Aoki&M.Yokoyama,Arch Androl 47,1-7(2001))。此外,雄激素与前列腺癌发生相关。雄激素在啮齿类动物模型中诱导前列腺癌发生(R.L.Noble,Cancer Res 37,1929-1933(1977);R.L.Noble,Oncology 34,138-141(1977))并且以合成代谢类固醇的形式接受雄激素的男性具有更高的前列腺癌发病率(J.T.Roberts&D.M.Essenhigh,Lancet 2,742(1986);J.A.Jackson,J.Waxman&A.M.Spiekerman,Arch Intern Med 149,2365-2366(1989);P.D.Guinan,W.Sadoughi,H.Alsheik,R.J.Ablin,D.Alrenga&I.M.Bush,Am J Surg 131,599-600(1976))。此外,如果人或狗在青春期前去势,则前列腺癌不会发展(J.D.Wilson&C.Roehrborn,J Clin Endocrinol Metab84,4324-4331(1999);G.Wilding,Cancer Surv 14,113-130(1992))。成年男性的去势导致前列腺退化和前列腺上皮凋亡(E.M.Bruckheimer&N.Kyprianou,Cell Tissue Res 301,153-162(2000);J.T.Isaacs,Prostate 5,545-557(1984))。这种对于雄激素的依赖性提供了用化学或手术去势(即雄激素除去)治疗前列腺癌的潜在理论基础。
前列腺癌症是西方国家中与男性癌症相关的第二位死亡导致原因(Damber,J.E.和Aus,G.Lancet(2008)371:1710-1721)。大量研究已经证实雄激素受体(AR)不仅对于前列腺癌发生是关键,而且使该病发展成去势抗性状态(Taplin,M.E.等人J.Clin.Oncol.(2003)21:2673-8;和Tilley,W.D.等人Cancer Res.(1994)54:4096-4102)。因此,有效抑制人AR保持对于治疗完全转移性前列腺癌的最有效的治疗方法之一。
AR具有所有核受体的模块化组成特征。它由N-末端结构域(NTD)、中心DNA结合结构域(DBD)、段铰链区和C-末端结构域构成,所述C-末端结构域包含激素配体结合袋(配体结合结构域,其还包含激素结合位点(HBS))和活化功能-2(AF2)位点(Gao,W.Q.等人Chem.Rev.(2005)105:3352-3370)。后者代表以正电荷和负电荷区为侧翼的AR表面上的疏水性沟-“电荷夹”,它们对于结合AR活化因子而言是有效的(Zhou,X.E.等人J.Biol.Chem.(2010)285:9161-9171)。近期研究已经鉴定了在AR上称作结合功能3(BF3)的新位点,其牵涉AR转录活性。当AR被易位入核时,DBD二聚化并且结合雄激素效应元件(AREs),且由此诱导转录,这是对于野生型AR和AR剪接变体的AR转录的必需过程。重要地,结合AREs的AR DBD二聚体的晶体结构是可得到的,这启示鉴定具有通过经基于合理的结构的药物设计靶向AR DBD的新机制的小分子抑制剂的可能性和易操作性。此外,因为迄今为止研究的所有抗性机制仍然牵涉AR结合至DNA且DBD以野生型AR和剪接变体存在,所以靶向DBD代表了克服抗性的新方法。
活化AR遵循得到充分表征的途径:在细胞质中,该受体结合维持AR激动剂结合构象的伴侣蛋白质(Georget,V.等人Biochemistry(2002)41:11824-11831)。在结合雄激素时,AR进行一系列的构象改变:从伴侣蛋白中解离、二聚化并且易位入核(Fang,Y.F.等人J.Biol.Chem.(1996)271:28697-28702;和Wong,C.I.等人J.Biol.Chem.(1993)268:19004-19012),其中它进一步与AF2位点上的辅激活因子蛋白发生相互作用(Zhou,X.E.等人J.Biol.Chem.(2010)285:9161-9171)。该事件触发RNA聚合酶II和其它因子募集以便与AR形成功能转录复合物。
值得注意地,目前的抗雄激素药例如比卡鲁胺、氟他胺、尼鲁米特和MDV3100均靶向这种特定过程。这些抗雄激素药通过结合AR配体结合位点起作用。因此,通过防止雄激素结合,它们还防止了对于辅激活因子相互作用所必需的受体的构象改变。尽管用这些AR抑制剂治疗最初可以抑制前列腺癌生长,但是长期激素疗法逐渐变成有效性降低(Taplin,M.E.等人J.Clin.Oncol.(2003)21:2673-8;和Tilley,W.D.等人Cancer Res.(1994)54:4096-4102)。因此,对于用于治疗癌症的靶向AR的另外的化合物存在显著性的需求。
雄激素还在女性癌症中发挥作用。一个实例是卵巢癌,其中升高水平的雄激素与发展卵巢癌的增加风险相关(K.J.Helzlsouer等人,JAMA 274,1926-1930(1995);R.J.Edmondson等人,Br J Cancer 86,879-885(2002))。已经在大多数卵巢癌中检测到AR(H.A.Risch,J Natl Cancer Inst 90,1774-1786(1998);B.R.Rao&B.J.Slotman,Endocr Rev 12,14-26(1991);G.M.Clinton&W.Hua,Crit Rev Oncol Hematol 25,1-9(1997)),而雌激素受体-α(ERa)和孕酮受体在少于50%的卵巢肿瘤中检测到。
概述
本发明部分基于如下令人意外的发现:本文所述的化合物调节雄激素受体(AR)活性。具体地,本文鉴定的化合物显示调节雄激素受体DNA-结合结构域(DBD)。
根据一个实施方案,提供具有式I的结构的化合物,其中
A可以是
R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3、NHCH3、CN或CF3;R2可以是H、CF3、OH、CH3、CN、NH2、CH2OH、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、CH2CH3或OCH2CH3;R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一;R3可以是H、F、CN、Cl、OH、SCH3、OCH3、O(CH2)2OCH3、CH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2OH、CH2CH3、OCH2CH3或CF3;R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;R5可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;G1可以是Br、Cl、I、CH3、H、F或OH;G2可以是Br、Cl、H、I、CH3、F或OH;G3可以是Cl、H、CH3、Br、I、F或OH;G4可以是Cl、H、Br、I、F或OH;G4可以任选地是CH3,条件是G3和G5不均是H;G5可以是H、CH2OH、Cl、Br、I、F或OH;G6可以是H、CH2OH、Cl、Br、I、F或OH;G7可以是H、CH2OH、Cl、Br、I、F或OH;G8可以是H、CH2OH、Cl、Br、I、F或OH;G9可以是H、CH2OH、Cl、Br、I、F或OH;G10可以是H、CH2OH、Cl、Br、I、F或OH;G11-G17独立地选自H、CH2OH、Cl、Br、I、F或OH;G18-G21独立地选自H、CH2OH、Cl、Br、I、F或OH;G22-G25独立地选自H、CH2OH、Cl、Br、I、F或OH;G26可以是H、CH2OH、Cl、Br、I、F或OH;G27可以是H、CH2OH、Cl、Br、I、F或OH;G28可以是H、CH2OH、Cl、Br、I、F或OH;G29可以是H、CH2OH、Cl、Br、I、F或OH;D可以是D可以任选地是条件是A是D可以任选地是条件是R1-R5不全部是H;J1可以是H、CH2CH3、CH3、Cl、Br、I、F、COOH或OH;J1可以任选地是CH3,条件是R1可以不是OH;J2可以是H、CH3、CH2CH3、Cl、Br、I、F或OH;J3可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J4可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J5可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J6可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J7可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J8可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J9可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J10可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J11可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J12可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J13可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;E可以是L1-L8可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L9-L16可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L17-L26可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L27-L30可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L31-L35可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L36-L39可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L40-L44可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L45-L47可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L48-L58可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L59可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L60可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L61可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L62可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L63可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L64可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L65可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L66可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;条件是该化合物不是如下化合物中的一个或多个:
根据另一个实施方案,提供调节AR活性的方法,该方法包括对有此需要的受试者施用具有式I的结构的化合物:其中A可以R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3、NHCH3、CN或CF3;R2可以是H、CF3、OH、CH3、CN、NH2、CH2OH、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、CH2CH3或OCH2CH3;R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一;R3可以是H、F、CN、Cl、OH、SCH3、OCH3、O(CH2)2OCH3、CH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2OH、CH2CH3、OCH2CH3或CF3;R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;R5可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;G1可以是Br、Cl、I、CH3、H、F或OH;G2可以是Br、Cl、H、I、CH3、F或OH;G3可以是Cl、H、CH3、Br、I、F或OH;G4可以是Cl、H、Br、I、F或OH;G4可以任选地是CH3,条件是G3和G5不均是H;G5可以是H、CH2OH、Cl、Br、I、F或OH;G6可以是H、CH2OH、Cl、Br、I、F或OH;G7可以是H、CH2OH、Cl、Br、I、F或OH;G8可以是H、CH2OH、Cl、Br、I、F或OH;G9可以是H、CH2OH、Cl、Br、I、F或OH;G10可以是H、CH2OH、Cl、Br、I、F或OH;G11-G17独立地选自H、CH2OH、Cl、Br、I、F或OH;G18-G21独立地选自H、CH2OH、Cl、Br、I、F或OH;G22-G25可以任选地选自H、CH2OH、Cl、Br、I、F或OH;G26可以是H、CH2OH、Cl、Br、I、F或OH;G27可以是H、CH2OH、Cl、Br、I、F或OH;G28可以是H、CH2OH、Cl、Br、I、F或OH;G29可以是H、CH2OH、Cl、Br、I、F或OH;D可以是D可以任选地是条件是A可以是D可以任选地是条件是R1-R5不全部是H;J1可以是H、CH2CH3、CH3、Cl、Br、I、F、COOH或OH;J1可以任选地是CH3,条件是R1可以不是OH;J2可以是H、CH3、CH2CH3、Cl、Br、I、F或OH;J3可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J4可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J5可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J6可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J7可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J8可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J9可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J10可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J11可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J12可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J13可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;E可以是L1-L8可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L9-L16可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L17-L26可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L27-L30可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L31-L35可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L36-L39可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L40-L44可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L45-L47可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L48-L58可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L59可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L60可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L61可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L62可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L63可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L64可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L65可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L66可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3。
根据另一个实施方案,提供本文所述的化合物在制备用于调节AR活性的药剂中的用途。
根据另一个实施方案,提供本文所述的权利要求11-21的化合物在调节AR活性中的用途。
根据另一个实施方案,提供本文所述的用于调节AR活性的化合物。
根据另一个实施方案,提供药物组合物,其包含本文所述的化合物和药学上可接受的载体或赋形剂。
根据另一个实施方案,提供商品包装,其包含本文所述的化合物和用于调节AR活性的说明书。
根据另一个实施方案,提供具有式II的结构的化合物,其中Q可以是M1可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;M2可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;M3可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;且M4可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;
条件是该化合物不是表1系列2部分中的化合物中除了化合物14409之外的一个或多个。该化合物可以是
根据另一个实施方案,提供调节AR活性的方法,该方法包括对有此需要的受试者施用具有式的结构II的化合物:其中Q可以是M1可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;M2可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;M3可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2;且M4可以是H、CH3、F、SO2CH3、OCH3、OH、NO2或NH2。
根据另一个实施方案,提供调节AR活性的方法,该方法包括对有此需要的受试者施用表1-系列3-6中列出的化合物。
根据另一个实施方案,提供本文所述的化合物在制备用于调节AR活性的药剂中的用途。
根据另一个实施方案,提供一组用于调节AR活性的本文所述的化合物。
根据另一个实施方案,提供药物组合物,该药物组合物包含本文所述的化合物和药学上可接受的载体或赋形剂。
根据另一个实施方案,提供商品包装,其包含本文所述的化合物和用于调节AR活性的说明书。
根据另一个实施方案,提供表1-系列3-6中列出的化合物在制备用于调节AR活性的药剂中的用途。
根据另一个实施方案,提供表1-系列3-6中列出的化合物在调节AR活性中的用途。
根据另一个实施方案,提供药物组合物,其包含如表1-系列3-6中列出的化合物和药学上可接受的载体或赋形剂。
根据另一个实施方案,提供商品包装,其包含如表1-系列3-6中列出的化合物和用于调节AR活性的说明书。
A可以是R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3、NHCH3、CN或CF3;R2可以是H、CF3、OH、CH3、CN、NH2、CH2OH、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、CH2CH3或OCH2CH3;R2可以任选地选自F、Cl、Br和I,条件是R1不是Cl、F、Br或I之一;R3可以是H、F、CN、Cl、OH、SCH3、OCH3、O(CH2)2OCH3、CH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2OH、CH2CH3、OCH2CH3或CF3;R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;R5可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3;G1可以是Br、Cl、I、CH3、H、F或OH;G2可以是Br、Cl、H、I、CH3、F或OH;G3可以是Cl、H、CH3、Br、I、F或OH;G4可以是Cl、H、Br、I、F或OH;G4可以任选地是CH3,条件是G3和G5不均是H;G5可以是H、CH2OH、Cl、Br、I、F或OH;G6可以是H、CH2OH、Cl、Br、I、F或OH;G7可以是H、CH2OH、Cl、Br、I、F或OH;G8可以是H、CH2OH、Cl、Br、I、F或OH;G9可以是H、CH2OH、Cl、Br、I、F或OH;G10可以是H、CH2OH、Cl、Br、I、F或OH;D可以是D可以任选地是条件是A是D可以任选地是条件是R1-R5不全部是H;J1可以是H、CH2CH3、CH3、Cl、Br、I、F、COOH或OH;J1可以任选地是CH3,条件是R1可以不是OH;J2可以是H、CH3、CH2CH3、Cl、Br、I、F或OH;J3可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J4可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J5可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J6可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J7可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J8可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J9可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J10可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J11可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J12可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;J13可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH;E可以是L1可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L2可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L3可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L4可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L5可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L6可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;L7可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3;且L8可以是H、CN、NH2、NO2、CH3、CH2CH3、Cl、Br、I、F、OH或CF3。
A可以是
R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3、NHCH3、CN或CF3。R2可以是H、CF3、OH、CH3、CN、NH2、CH2OH、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、CH2CH3或OCH2CH3。R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一。R3可以是H、F、CN、Cl、OH、SCH3、OCH3、O(CH2)2OCH3、CH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2OH、CH2CH3、OCH2CH3或CF3。R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3。R5可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3。G1可以是Br、Cl、I、CH3、H、F或OH。G2可以是Br、Cl、H、I、CH3、F或OH。G3可以是Cl、H、CH3、Br、I、F或OH。G4可以是Cl、H、Br、I、F或OH。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H、CH2OH、Cl、Br、I、F或OH。D可以是D可以任选地是条件是A是D可以任选地是条件是R1-R5不全部是H。J1可以是H、CH2CH3、CH3、Cl、Br、I、F或OH。J1可以任选地是CH3,条件是R1可以不是OH。J2可以是H、CH3、CH2CH3、Cl、Br、I、F或OH。J3可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J4可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J5可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J6可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J7可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J8可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J9可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。E可以是A可以是R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3、NHCH3、CN或CF3。R2可以是H、CF3、OH、CH3、NH2、CN、CH2OH、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、CH2CH3或OCH2CH3。R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一。R3可以是H、F、CN、Cl、OH、SCH3、OCH3、O(CH2)2OCH3、CH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2OH、CH2CH3、OCH2CH3或CF3。R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3。R5可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、O(CH2)2OCH3、NH2、SO2CH3、OCH(CH3)2、NHCH3、Br、I、CH2CH3、OCH2CH3或CF3。G1可以是Br、Cl、I、CH3、H、F或OH。G2可以是Br、Cl、H、I、CH3、F或OH。G3可以是Cl、H、CH3、Br、I、F或OH。G4可以是Cl、H、Br、I、F或OH。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H、CH2OH、Cl、Br、I、F或OH。D可以是J1可以是H、CH2CH3、CH3、Cl、Br、I、F或OH。J1可以任选地是CH3,条件是R1可以不是OH。J2可以是H、CH3、CH2CH3、Cl、Br、I、F或OH。J3可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。J4可以是H、CN、CH3、CH2CH3、Cl、Br、I、F或OH。E可以是A可以是R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3或CF3。R2可以是H、CF3、OH、CN、CH3或NH2。R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一。R3可以是H、F、CN、Cl、OH、SCH3、OCH3、CH3、NH2、Br、I、CH2OH、CH2CH3或CF3。R4可以是H、CH3、NHCH3、OH、CH2OH、F、CN、Cl、SCH3、OCH3、NH2、Br、I或CF3。R5可以是H、CH3、OH、F、CN、Cl、NH2、Br、I或CF3。G1可以是Br、Cl、I、CH3、H、F或OH。G2可以是Br、Cl、H、I、CH3、F或OH。G3可以是Cl、H、CH3、Br、I、F或OH。G4可以是Cl、H、Br、I、F或OH。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H、CH2OH、Cl、Br、I、F或OH。D可以是J1可以是H、CH2CH3、CH3、Cl、Br、I、F或OH。J1可以任选地是CH3,条件是R1可以不是OH。J2可以是H、CH3、Cl、Br、I、F或OH。J3可以是H、CN、CH3、Cl、Br、I、F或OH。J4可以是H、CN、CH3、Cl、Br、I、F或OH。E可以是A可以是R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2、O(CH2)2OCH3、Br、I、CN、CH2OH、CH2CH3、OCH2CH3或CF3。R2可以是H、CF3、OH、CN、CH3或NH2。R2可以任选地选自F、Cl、Br和I,条件是R1可以不是Cl、F、Br或I之一。R3可以是H、F、CN、Cl、OH、SCH3、OCH3、CH3、NH2、Br、I或CF3。R4可以是H、CH3、NHCH3、OH、CH2OH、F、Cl、OCH3、NH2、Br、I或CF3。R5可以是H、CH3、OH、F、CN、Cl、NH2、Br、I或CF3。G1可以是Br、Cl、I、CH3、H、F或OH。G2可以是Br、Cl、H、I、CH3、F或OH。G3可以是Cl、H、CH3、Br、I、F或OH。G4可以是Cl、H、Br、I、F或OH。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H、CH2OH、Cl、Br、I、F或OH。D可以是J1可以是H、CH2CH3、CH3、Cl、Br、I、F或OH。J1可以任选地是CH3,条件是R1可以不是OH。J2可以是H、CH3、Cl、Br、I、F或OH。J3可以是H、CN、CH3、Cl、Br、I、F或OH。J4可以是H、CN、CH3、Cl、Br、I、F或OH。E可以是A可以是R1可以是H、OCH3、OH、CH3、NH2、Cl、SO2CH3、OCH(CH3)2或O(CH2)2OCH3。R2可以是H或CF3。R2可以任选地选自F和Cl,条件是R1不是Cl、F、Br或I之一。R3可以是H、F、CN、Cl、OH或SCH3。R4可以是H、CH3、NHCH3、OH、CH2OH或F。R5可以是H。G1可以是Br、Cl、I或CH3。G2可以是Br、Cl、H、I或CH3。G3可以是Cl、H、CH3或Br。G4可以是Cl、H或Br。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H或CH2OH。D可以是J1可以是H或CH2CH3。J1可以任选地是CH3,条件是R1可以不是OH。J2可以是H。J3可以是H或CN。J4可以是H。E可以是A可以是R1可以是H、OCH3、OH或CH3。R2可以是H或CF3。R2可以任选地选自F和Cl,条件是R1可以不是Cl、F、Br或I之一。R3可以是H或F。R4可以是H或CH3。R5可以是H。G1可以是Br或Cl。G2可以是Br、Cl或H。G3可以是Cl、H、CH3或Br。G4可以是Cl、H或Br。G4可以任选地是CH3,条件是G3和G5不均是H。G5可以是H或CH2OH。D可以是J1可以是H。J2可以是H。J3可以是H。J4可以是H。E可以是
该化合物可以选自如下化合物中的一个或多个:
该化合物可以选自如下化合物中的一个或多个:
该化合物可以选自如下化合物中的一个或多个:
该化合物可以选自如下化合物中的一个或多个:
该化合物可以选自如下化合物中的一个或多个:
调节AR活性可以用于治疗选自如下的至少一种适应证:癌症、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和与年龄相关的黄斑变性。所述癌症可以是AR-介导的癌症。所述癌症可以选自:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌和膀胱癌。所述癌症可以是紫杉烯(Taxene)抗性(resistant)三阴性乳腺癌。
根据一个实施方案,提供具有选自表1或权利要求中所述的结构的化合物或其药学上可接受的盐的用途。
根据另一个实施方案,提供具有选自表1或权利要求中所述的结构的化合物或其药学上可接受的盐的用途。
根据另一个实施方案,提供药物组合物,其包含本文举出的化合物或其药学上可接受的盐和药学上可接受的赋形剂。
根据另一个实施方案,提供用于调节AR活性的方法,该方法包括对哺乳动物细胞施用本文举出的化合物或其药学上可接受的盐。
根据另一个实施方案,提供用于调节雄激素受体(AR)活性的药物组合物,其包含如本文所述的化合物和药学上可接受的载体。
根据另一个实施方案,提供商品包装,其包含(a)本文所述的化合物或本文所述的药物组合物;和(b)其用于调节雄激素受体(AR)活性的使用说明书。
调节AR活性可以用于治疗选自如下的至少一种适应证:癌症、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和与年龄相关的黄斑变性。所述癌症可以是AR-介导的癌症。所述癌症可以选自:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌和膀胱癌。所述癌症可以是紫杉烯抗性三阴性乳腺癌。调节AR活性可以用于治疗前列腺癌。
所述哺乳动物细胞可以是人细胞。该细胞可以是前列腺细胞。该细胞可以是前列腺癌细胞。
根据另一个实施方案,提供具有选自表1或权利要求中所述的结构的化合物。
所述化合物可以用于调节雄激素受体(AR)活性。调节雄激素受体(AR)活性可以用于治疗选自如下的至少一种适应证:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和与年龄相关的黄斑变性。调节AR活性可以用于治疗选自如下的至少一种适应证:癌症、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和与年龄相关的黄斑变性。所述癌症可以是AR-介导的癌症。所述癌症可以选自:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌和膀胱癌。所述癌症可以是紫杉烯抗性三阴性乳腺癌。调节AR活性可以用于治疗前列腺癌。
附图简述
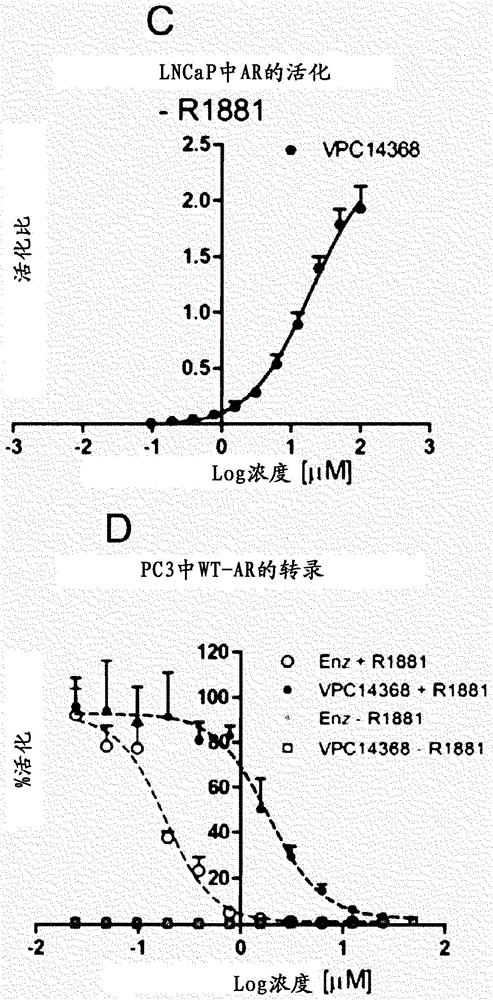
图1显示剂量响应曲线(0-12.5μM),其示例使用eGFP(A)和PSA(B)测定法、在R1881的存在下14368对LNCaP细胞中AR转录活性的抑制。(C)无R1881存在下14368对LNCaP细胞中的AR转录的活化。值1相当于来自用0.1nM R1881刺激的LNCAP细胞的eGFP信号。显示用WT-AR(D)或T877A-AR突变体(E)瞬时转染的PC3细胞中AR转录活性的荧光素酶报道分子。用14368或恩杂鲁胺(Enzalutamide)(Enz)在R1881刺激的存在下无其刺激的存在下处理细胞。数据点表示三次独立实验的平均值±SEM。100%是指仅用0.1%DMSO记录的发光。(F)14368和比卡鲁胺(Bic)在0.1nM R1881的存在下对R1-AD1细胞中内源性野生型AR的抑制。
图2左侧小组:14368、14435、14436、14439、14404和比卡鲁胺(Bic)、使用eGFP AR转录测定法、在0.1nM R1881的存在下对AR转录抑制的剂量响应曲线。右侧小组:用14368、14435、14436、14439和14404、使用极性筛选雄激素受体竞争绿色测定试剂盒的雄激素置换的剂量响应曲线。
图3显示14228的体外特性,包括eGFP转录活性、PSA、细胞存活率、雄激素置换和BLI。
图4对AR突变体(Glu592、Tyr594、Val582、Arg586和Phe583)与野生型AR的14228的转录活性比较。
图5显示假拟原子所示的人AR DBD同源性模型上预计的结合位点的图示。该位点被人AR DBD中的残基Ser579、Val582、Phe583、Arg586、Gln592、Tyr594、Arg609、Lys610和Pro613包被,其中阴影区位于Phe583、Tyr594、Lys610与Pro613之间,相邻Val582和Lys610右侧代表疏水区。此外,阴影区Arg586、Arg609、Arg616和Lys610代表极性区。
图6显示对AR具有特异性的DBD-相互作用化合物的选择的棒形图,其中在对瞬时表达的AR、GR和PR或对MCF-7细胞中内源性ER-_的荧光素酶测定中以所示浓度测试的恩杂鲁胺(enz)在示意图(A)、14228(B)和14449(C)中显示,并且使用ARR3tk-荧光素酶报道分子评价AR、GR和PR活性。MCF-7细胞包括稳定转染的雌激素响应元件-荧光素酶基因。100%是指仅使用有0.1%DMSO的各受体的荧光素酶活性(误差条表示平均值_S.D.,6次重复enz,恩杂鲁胺)。
图7显示DBD-相互作用化合物对AR靶基因表达的作用,A是显示从LNCaP细胞中分泌的PSA的线性图,所述LNCaP细胞用1nM R1881和化合物(即恩杂鲁胺、14449和14228)以所示浓度处理2天,其中分泌的PSA通过分析来此2次独立实验的每个孔的150μl细胞培养物来定量。B是显示在R1881、化合物14449和恩杂鲁胺(Enz)的存在下AR靶基因和非雄激素响应基因(α-肌动蛋白,ACTB)的基因表达改变的棒形图*=基于14449+R1881与DMSO+R1881和恩杂鲁胺+R1881与DMSO+R1881之间的2-样品t检验的基因表达显著性下降(p值<0.05)。
图8显示化合物-14449减小肿瘤体积并且消除去势小鼠的LNCaP异种移植物模型中的PSA产生,对所述小鼠每日2次施用14449(100mg/kg)或恩杂鲁胺(Enz)(10mg/kg)4周并且评价LNCaP异种移植物肿瘤体积(A)和血清PSA(B),其中将数据表示为平均值±S.E.,n=4。P值<0.05被视为与媒介物对照组相比具有显著性(*);p值<0.001被视为与媒介物对照组相比具有极其的显著性(**)。
图9显示一系列示意图,(A)使用LNCaP eGFP细胞在0.1nM R1881的存在下,通过测定荧光来比较化合物25(即14449)与恩杂鲁胺的AR抑制活性;(B)通过使用相同的LNCaP eGFP细胞测定分泌入培养基的PSA来评价中心化合物的抑制作用;和(C)使用MTS测定法的25(即14449)对LNCaP、MR49F(恩杂鲁胺-抗性)和PC3细胞的抗增殖作用,其中用不同浓度的所述抑制剂在0.1nM R1881的存在下将LNCaP、MR49F和PC3细胞处理3天。
详细描述
DNA-结合结构域是用于抑制AR二聚化和/或DNA结合的有吸引力的靶标。生物信息学(In silico)计算机药物研发方法用于进行>3百万可购买到的来自ZINC数据库的先导类化合物的实际筛选(Irwin,J.等人Abstracts of Papers Am.Chem.Soc.(2005)230:U1009),以鉴定潜在的DBD结合物。生物信息学方法包括大规模对接、原位重新评分(in-site rescoring)和共享投票(consensus voting)法。
本领域技术人员应当理解,COOH和NR2可以包括相应的离子,例如分别为羧酸根离子和铵离子。或者,如果显示离子,则本领域技术人员可以理解还可以存在抗衡离子。此外,本领域技术人员应当理解,其它部分可以包括相应的离子,且如果显示离子,则本领域技术人员可以理解还可以存在抗衡离子。
表1显示根据结构(系列1-6)和相关标识符测试的化合物。表1还提供了鉴定本发明范围内的化合物的另外的一般结构。阴影化合物是新的合成化合物,而无阴影化合物是已知的ZINC化合物。此外,如果其%抑制、eGFP IC50或PSA IC50不具有指定的值,则这可以归因于在%抑制的情况中未取测量值或未计算该值。因此,表1中没有给出值并不意味着在不存在活性。此外,对于那9种新化合物(14502-14510)的0值,未如使用其它化合物所进行的进行单一浓度筛选。而是直接使用多个浓度测定IC50,且对于这些无活性化合物,剂量响应曲线是平坦的,显示在所有浓度下几乎为0%抑制。
对于其它化合物,对于它们中的大部分进行单一剂量筛选,而对于具有高抑制百分比的那些仅测定IC50,除了少量具有相对低%抑制的有意义的化合物之外。当以3μM或1μM浓度测定%抑制时,对于活性和无活性化合物的%抑制阈值低至你所认可的程度。发现在3μM下约30%的抑制率可以得到约50μM的IC50。因此,一般具有低于30%的抑制值的化合物可以被视为无活性的。尽管如此,但是化合物的活性不应当始终根据%抑制来确定。实际上,如果系统的敏感性在一定程度上下降,则所测试和视为无活性的任意化合物都可以被视为有活性。当化合物变得越来越好时,敏感性水平增加。因此,在测定系统中测定低活性化合物变得更为困难。然而,由于较新的化合物基于上述成功的化合物,所以它们在一些水平上可以具有一定的活性。因此,“无活性”可能是不正确的术语,而“未检测到”可能更适合。
本领域技术人员将理解,所述部分与如本文所述的化合物的共价连接点可以是,例如、但不限于在特定条件下裂解。特定条件可以包括,例如、但不限于体内酶或非酶方式。所述部分的裂解可以例如、但不限于自发地进行或可以被催化、被另一种试剂或物理参数或环境参数的改变诱导,例如酶、光、酸、温度或pH。所述部分可以是,例如、但不限于起掩蔽官能团作用的保护基、作为一种或多种主动或被动转运机制的底物起作用的基团或起传递或增强化合物特性例如溶解性、生物利用度或定位的基团。
在一些实施方案中,如上述表1和权利要求中所述的化合物可以用于至少一种适应证的全身治疗,所述至少一种适应证选自:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和老年性黄斑退化症。在一些实施方案中,表1和如权利要求中所述的化合物可以用于制备用于全身治疗本文所述的适应证的药剂或组合物。在一些实施方案中,还提供了全身治疗本文所述的任意适应证的方法。
本文所述的化合物可以呈游离形式或其盐的形式。在一些实施方案中,本文所述的化合物可以呈药学上可接受的盐的形式,其在本领域中是已知的(Berge S.M.等人,J.Pharm.Sci.(1977)66(1):1-19)。本文所使用的药学上可接受的盐包括,例如,具有母体化合物的所需药理学活性的盐(保持母体化合物的生物学效能和/或性质并且在生物学和/或其它方面不是不合需要的盐)。具有一个或多个能够成盐的官能团的本文所述的化合物可以例如形成为药学上可接受的盐。含有一个或多个碱性官能团的化合物可能能够与例如药学上可接受的有机酸或无机酸形成药学上可接受的盐。药学上可接受的盐可衍生自,例如但不限于,乙酸、己二酸、藻酸、天冬氨酸、抗坏血酸、苯甲酸、苯磺酸、丁酸、肉桂酸、柠檬酸、樟脑酸、樟脑磺酸、环戊烷丙酸、二乙基乙酸、二葡糖酸、十二烷基磺酸、乙磺酸、甲酸、富马酸、葡庚糖酸、葡糖酸、甘油磷酸、乙醇酸、半磺酸、庚酸、己酸、盐酸、氢溴酸、氢碘酸、2-羟基乙磺酸、异烟酸、乳酸、苹果酸、马来酸、丙二酸、扁桃酸、甲磺酸、2-萘磺酸、萘二磺酸、对甲苯磺酸、烟酸、硝酸、草酸、扑酸、果胶酸、3-苯基丙酸、磷酸、苦味酸、庚二酸、特戊酸、丙酸、丙酮酸、水杨酸、琥珀酸、硫酸、氨基磺酸、酒石酸、硫氰酸或十一烷酸。含有一个或多个酸性官能团的化合物可能能够与例如药学上可接受的碱形成药学上可接受的盐,所述药学上可接受的碱例如但不限于,基于碱金属或碱土金属的无机碱,或例如伯胺化合物、仲胺化合物、叔胺化合物、季胺化合物、取代胺、天然存在的取代胺、环状胺或碱性离子交换树脂的有机碱。药学上可接受的盐可衍生自,例如但不限于,药学上可接受的金属阳离子例如铵、钠、钾、锂、钙、镁、铁、锌、铜、锰或铝的氢氧化物、碳酸盐或碳酸氢盐、氨、苄星青霉素、葡甲胺、甲基胺、二甲胺、三甲胺、乙基胺、二乙胺、三乙胺、异丙胺、三丙胺、三丁基胺、乙醇胺、二乙醇胺、2-二甲基氨基乙醇、2-二乙基氨基乙醇、二环己基胺、赖氨酸、精氨酸、组氨酸、咖啡因、哈胺、胆碱、甜菜碱、乙二胺、氨基葡萄糖、葡糖胺、甲基葡糖胺、可可碱、嘌呤、哌嗪、哌啶、普鲁卡因、N-乙基哌啶、可可碱、四甲基铵化合物、四乙基铵化合物、吡啶、N,N-二甲基苯胺、N-甲基哌啶、吗啉、N-甲基吗啉、N-乙基吗啉、二环己基胺、二苄基胺、N,N-二苄基苯乙基胺、1-二苯羟甲胺(ephenamine)、N,N'-二苄基乙二胺或聚胺树脂。在一些实施方案中,本文所述的化合物可以含有酸性基团和碱性基团二者,并且可以呈内盐或两性离子形式,例如但不限于,甜菜碱。本文所述的盐可以通过本领域技术人员已知的常规方法制备,例如但不限于,通过将游离形式与有机酸或无机酸或碱反应,或通过从其它盐进行阴离子交换或阳离子交换。本领域技术人员将理解,盐的制备可以在分离和纯化化合物过程中原位进行,或者盐的制备可以通过单独分离和纯化的化合物反应来进行。
在一些实施方案中,本文所述的化合物及其所有不同形式(例如,游离形式、盐、多晶型物、异构形式)可以呈溶剂加合物形式,例如溶剂化物形式。溶剂化物含有化学计算或非化学计算量的与化合物或其盐物理缔合的溶剂。所述溶剂可以例如但不限于药学上可接受的溶剂。例如,当溶剂是水时形成水合物,或当溶剂是醇时形成醇化物。
在一些实施方案中,本文所述的化合物及其所有不同形式(例如,游离形式、盐、溶剂化物、异构形式)可以包括结晶和无定形形式,例如多晶型物,假多晶型物,构象多晶型物,无定形形式,或它们的组合。多晶型物包括相同元素组成的化合物的不同晶体堆积排列。多晶型物通常具有不同的X-射线衍射图、红外光谱、熔点、密度、硬度、晶形、光学和电学性质、稳定性和/或溶解性。本领域技术人员将理解,包括重结晶溶剂、结晶速率和贮存温度的各种因素可以导致单一晶型占优势。
在一些实施方案中,本文所述的化合物及其所有不同形式(例如,游离形式、盐、溶剂化物、异构形式)包括异构体,例如几何异构体、基于不对称碳的旋光异构体、立体异构体、互变异构体、单一对映异构体、单一非异构对映异构体、外消旋体、非对映异构体混合物、和它们的组合,并且不限于为了便利目的而示例的结构的描述。
在一些实施方案中,化合物可以包括类似物、异构体、立体异构体或相关衍生物。本发明的化合物可以包括涉及表1的化合物的化合物,通过用紧密相关的取代基取代或替代一些取代基例如用相关卤素替代卤素取代基(即溴取代氯等)或用不同长度的相关烷基链替代烷基链等来进行。在另外的实施方案中,化合物可以包括具有一般或马库什结构范围内的化合物,正如根据表1中呈现的数据中鉴定的结构-活性相关性所确定的。作为实例,环结构A、B和C具有不同的组成,视测试化合物的类型而定。已经显示出具有良好效能的不同环结构可以与其它有效的环结构合并,只要维持表1中所示的A-B-C构型。按照这种方式,可以预期任意不同的环结构组合也是有效的。测定这类结构-活性相关性以便研发一般的马库什结构属于本领域技术人员的范围。
在一些实施方案中,如本文所述的药物组合物可以包含这类化合物的盐,优选药学上或生理学上可接受的盐。药物制剂将典型地包含一种或多种载体、赋形剂或稀释剂,其适于制剂的施用模式、例如通过注射、吸入、局部施用、灌洗或适于所选治疗的其它模式。适合的载体、赋形剂或稀释剂(在本文中可以互换使用)是本领域已知用于这类施用模式的那些。
适合的药物组合物可以通过本领域已知的方法配制,并且它们的施用模式和剂量由熟练的专业人员确定。关于肠胃外施用,可以将化合物溶解在无菌水或盐水或药学上可接受的载体中,所述药用载体用于非水溶性化合物的施用,例如用于维生素K的那些。对于经肠施用,化合物可以是以片剂、胶囊或溶解在液体形式中施用。片剂或胶囊可以是肠包衣的,或者于持续释放的制剂中。许多适当的制剂是已知的,包括包封待释放的化合物的聚合物或蛋白微粒、软膏剂、糊剂、凝胶、水凝胶、或可以局部或区域使用以施用化合物的溶液。持续释放贴片或植入物可以用于提供延长的时段内的释放。许多本领域技术人员已知的技术描述在Remington:the Science&Practice of Pharmacy,Alfonso Gennaro,第20版,Lippencott Williams&Wilkins,(2000)中。用于肠胃外施用的制剂可以例如含有赋形剂、聚亚烷基二醇如聚乙二醇、植物来源的油或氢化萘。生物相容的、生物可降解的丙交酯聚合物、丙交酯/乙交酯共聚物、或聚氧乙烯-聚氧丙烯共聚物可以用于控制化合物的释放。其它潜在有用的用于调节化合物的肠胃外递送系统包括乙烯-醋酸乙烯酯共聚物颗粒、渗透泵、可植入输注系统和脂质体。用于吸入的制剂可以含有赋形剂例如乳糖,或者可以是含有例如聚氧乙烯-9-月桂醚、甘胆酸盐和去氧胆酸盐的水溶液,或者可以是用于以滴鼻剂或作为凝胶形式施用的油性溶液。
如本文所述或如本文所述应用的化合物或药物组合物可以通过医疗装置或器具施用,例如植入物、移植物、假体、支架等。此外,可以设计植入物,预期其可以包含和释放这类化合物或组合物。一个实例可以是由适合于在一定时间期限内释放化合物的聚合物材料构成的植入物。
如本文所述的药物组合物的“有效量”包括治疗有效量或预防有效量。“治疗有效量”是指在所需剂量下和时期内有效获得所需治疗结果(如减小的肿瘤尺寸、增加的寿命或增加的预期寿命)的量。化合物的治疗有效量可以根据例如以下因素改变:受试者的疾病状态、年龄、性别和体重以及化合物在受试者中引发所需反应的能力。剂量给药方案可以进行调整以提供最佳的治疗反应。治疗有效量也是其中化合物的任何毒性或有害作用被治疗有效作用超过的量。“预防有效量”是指在所需剂量下和时期内有效获得所需预防结果(例如较小的肿瘤,增加的寿命,增加的预期寿命或防止前列腺癌向雄激素非依赖性形式进展)的量。典型地,在疾病之前或疾病早期在受试者中使用预防剂量,因此预防有效量可以少于治疗有效量。
应当注意,剂量值可以随将要缓解的病症的严重性而变化。对于任何具体的受试者,具体的剂量给药方案可以随时间根据个体需要和施用组合物或监督组合物给药的人的专业判断进行调整。本文列出的剂量范围仅是例举性的,并且不限制医学从业者可以选择的剂量范围。在组合物中活性化合物的量可以按照例如以下因素而改变:受试者的疾病状态、年龄、性别和体重。剂量给药方案可以进行调整以提供最佳的治疗反应。例如,可以施用单次推注,可以随时间施用数个分剂量,或者剂量可以根据治疗情形的紧急情况要求而成比例的减少或增加。以单位剂量形式配制肠胃外组合物可以是有利的,以便易于施用和剂量一致性。
在一些实施方案中,本文所述的化合物及其所有不同形式可以非限制性地例如与用于至少一种选自以下组成的组的适应证的其它治疗方法联合使用:前列腺癌、乳腺癌、卵巢癌、子宫内膜癌、脱发、痤疮、多毛症、卵巢囊肿、多囊卵巢病、性早熟和与年龄相关的黄斑变性。例如,本文所述的化合物和它们所有的不同形式可以与外科手术、放射(近距放射疗法或外线束)或其它疗法(例如HIFU)一起作为新辅助疗法(之前)、附属疗法(期间)、和/或辅助性疗法(之后)使用。
通常,本文所述的化合物应当在不导致实质毒性情况下使用。本文所述化合物的毒性可以使用标准技术测定,例如通过在细胞培养物或实验动物中测试和确定治疗指数,即LD50(50%群体致死剂量)和LD100(100%群体致死剂量)之间的比率。然而,在一些情况下,例如在重病情况下,可能需要施用显著过量的组合物。本文所述的一些化合物可以在一些浓度下是有毒性的。可以使用滴定研究来测定毒性和无毒性浓度。可以如下评估毒性:使用不表达AR的PC3细胞作为阴性对照,研究具体化合物或组合物对细胞系的特异性。可以使用动物研究来提供证明化合物是否对于其它组织具有任何影响。靶向AR的系统治疗将不大可能对其它组织导致重大问题,因为抗雄激素药和雄激素不敏感综合征不是致命的。
本文所述的化合物可以施用于受试者。如本文所用,“受试者”可以是人、非人灵长类、大鼠、小鼠、牛、马、猪、绵羊、山羊、狗、猫等。受试者可以被猜测患有癌症或处于癌症(如前列腺癌,乳腺癌,卵巢癌或子宫内膜癌)风险下,或被怀疑患有或处于以下疾病风险下:痤疮、多毛症、脱发、良性前列腺肥大、卵巢囊肿、多囊卵巢病、性早熟或与年龄相关的黄斑变性。本领域普通技术人员已知用于各种癌症(例如前列腺癌、乳腺癌、卵巢癌或子宫内膜癌)的诊断方法和用于痤疮、多毛症、脱发、良性前列腺肥大、卵巢囊肿、多囊卵巢病、性早熟或与年龄相关的黄斑变性的诊断方法,和癌症(例如前列腺癌、乳腺癌、卵巢癌或子宫内膜癌)的临床叙述、痤疮、多毛症、脱发、良性前列腺肥大、卵巢囊肿、多囊卵巢病、性早熟或与年龄相关的黄斑变性的诊断和临床叙述。
所用的定义包括使用用于研究目的的雄激素例如双氢睾酮(DHT)或合成雄激素(R1881)对雄激素受体(AR)的配体依赖性活化。AR的配体依赖性活化是指在没有雄激素(配体)的存在下对AR的反式激活,例如,用福司柯林(FSK)刺激cAMP-依赖性蛋白激酶(PKA)途径。
如本文所述的一些化合物和组合物可以干扰特异性二聚化-和/或DNA-结合依赖性活化(例如潜在地结合AR DBD以通过破坏AR二聚化和防止DBD-DNA结合阻断AR转录的机制。
本发明的不同可选实施方案和实施例如本文所述。这些实施方案和实施例是示例性的,而不应当被视为限定本发明的范围。
材料和方法
生物信息学流水线
1.蛋白质和配体制备
使用Maestro 9.3(LLC)内的Protein Preparation Wizard制备AR DBD二聚体-DNA(1R4I.pdb)的晶体结构。加入氢原子,指定键级并且使用Prime添加用于一些残基的缺失侧链。使用OPLS-2005力场使侧链最小化。
将具有3百万个小分子的先导类ZINC试剂盒输入Molecular Operating Environment(MOE)2010。通过洗涤过程使全部分子质子化/脱质子化,添加偏好电荷并且用MMFF94x力场最小化至梯度。在最小化后,将数据库作为sdf文件输出。
2.AR DBD中的成药(Druggable)结合位点检测
为了鉴定AR DBD二聚体-DNA结构中潜在的成药结合位点,使用基于几何学和给予能量的方法如MOE 2010内的Site Finder、Pocket-Finder和Q-siteFinder检测AR DBD二聚体-DNA复合物、DBD二聚体和DBD单体。检查可能的结合位点并且基于参数如大小、形状、氨基酸组成和袋体积比较。
3.潜在AR DBD结合物的虚拟筛选
Maestro 9.3和eHiTs 2011中的两种对接程序Glide用于虚拟筛选。预测的结合位点中的残基用于确定虚拟筛选的活性位点。为了Glide对接,使用位于选择的残基中心的盒确定栅格。不应用系统规定参数,且将全部设置值保持为默认值。使用Glide SP模式对接ZINC数据库并且制定截止值以便清除对受体具有潜在低结合亲和力的化合物。使剩余的化合物进行默认设置中的eHiTs对接。eHiTs的截止值用于保持用两种对接程序良好地保持对化合物始终如一地评分。使用来自两种程序的对接方式计算RMSD(根中值平方差(root median square deviation))值,并且除去RMSD值高于的化合物。基于MOE 2010中的结构相似性集合其余化合物并且化合物根据与受体的有利相互作用选自上部有序的集合。
化合物的体外鉴定
1.细胞培养:LNCaP和PC3人前列腺癌细胞得自美国模式培养物保藏中心(American Type Culture Collection)(ATCC,Manassas,VA),并且使其生长在补充5%胎牛血清(FBS)(Invitrogen)的RPMI1640培养基中。使用慢病毒方法,用与eGFP报道基因(LN-ARR2PB-eGFP)融合的雄激素响应前列腺碱性蛋白衍生的启动子稳定转染LNCaP eGFP细胞系,并且使其生长在无酚红的补充了5%CSS的RPMI 1640中。在补充了5%FBS和10μM MDV3100的RPMI 1640中培养Inihouse研发的MDV3100-抗性LNCaP细胞。将全部细胞维持在37℃、5%CO2下。
2.eGFP细胞AR转录测定:如上所述测定AR转录活性(Tavassoli,Snoek等人2007)。
3.前列腺-特异性抗原(PSA)测定:使用相同板与eGFP测定平行地进行分泌入培养基的PSA水平的评价。在将细胞温育3天后,从每个孔中取出150μl培养基并且加入到150μl PBS中。然后使用Cobas e 411分析仪(Roche Diagnostics)、根据制造商的说明书评价PSA水平。
4.生物层干扰量度(BLI)测定:如上所述测定小分子与AR之间的直接可逆相互作用(Lack,Axerio-Cilies等人2011)。
5.雄激素置换测定:使用极性筛选雄激素受体竞争物绿色测定试剂盒(Polar Screen Androgen Receptor Competitor Green Assay Kit)、根据制造商的说明书评价雄激素置换(Lack,Axerio-Cilies等人2011)。
6.SRC2-3肽置换测定:如上所述测定AR AF2特异性肽置换。
7.细胞存活测定:将PC3、LNCaP和MDV3100-抗性细胞以3,000细胞/孔在96-孔板中的包含5%活性炭处理的血清(charcoal stripped serum)(CSS)的RPMI 1640上PC3铺板,用0.1nM R1881和化合物(0-25μM)处理96hrs。在4天处理后,使用3-(4,5-二甲基噻唑-2-基)-5-(3-羧基甲氧基苯基)-2-(4-磺苯基)-2H-四唑鎓测定法、根据制造商的方案测定细胞密度(CellTiter 961 Aqueous One Solution Reagent,Promega)。
8.突变研究:使用QuickchangeTM定点诱变试剂盒、根据说明书使预测结合位点上的残基突变。
9.瞬时转染:将PC3/MCF7细胞接种入96-孔板(2,000细胞/孔)。24hrs后,使用转染试剂TT20将野生型AR(50ng/孔)/AR突变体和AR3TK-荧光素每日质粒(1:3)共转染入PC3细胞或将ERE-荧光素酶(50ng/孔)转染入MCF7细胞。转染后24hrs,用不同浓度的化合物处理细胞,并且在24hrs后,裂解细胞,且使用发光计取读数值。
构建体
在pcDNA3.1表达质粒上编码全长人AR(hARWT)或剪接变体(AR-V7)。
使用QuikChangeTM诱变试剂盒(StratageneTM)、应用hARWT或AR-V7模板生成DBD中的点突变。使用引物设计工具(AgilentTM)生成诱变引。如上所述(Miesfeld,R.等人,(1986)Genetic complementation of a glucocorticoid receptor deficiency by expression of cloned receptor cDNA.Cell 46,389-399)从pGR哺乳动物表达载体中表达糖皮质激素受体(GR)。从pSG5-PRB载体中表达孕激素受体(PR)且得自Dr.X.Dong。从hARWT构建体中扩增AR-DBD_铰链结构域(氨基酸558-689)并且使用聚合酶不完全引物延伸法(PIPE)克隆入pTrc表达载体(N-末端His6标记,InvitrogenTM)。简言之,通过混合得自如下模板和引物的PCR产物克隆AR-DBD+铰链结构域:来自hARWT模板5'-CAT CAT CAT CAT CAT CAT GGT ACC TGC CTGATCTGT GG和5'-CAG GCT GAA AAT CTT CTC TCA GTGTCC AGC ACACAC TAC AC的AR(558-689)插入物;缺乏多克隆位点5'-ATCTCCACAGATCAGGCAGGT ACCATGATG ATG ATG ATG ATG和5'-GGT GTA GTG TGT GCTGGA CAC-TGA GAG AAG ATT TTC AGC CTG的pTrc载体;加下划线的引物片段退火至特定模板,不过,其5'-延伸部分(用斜体字表示)与另一PCR的相应的引物序列互补。通过混合来自每次反应的PCR产物进行质粒装配(Klock,H.E.等人,(2008)Combining the polymerase incomplete primer extension method for cloning and mutagenesis with microscreening to accelerate structural genomics efforts.Proteins 71,982-994),然后转入化学感受态细菌。类似策略用于将AR-DBD+铰链克隆入表达N-末端生物素化序列(GLNDIFEAQKIEWHE)和C-末端His标记的Pan4载体(亲和力)。YFP-AR质粒为来自Dr.Jan Trapman的赠品(van Royen,M.E.等人,(2012)Stepwise androgen receptor dimerization.J.Cell Sci.125,1970-1979)且基于pEYFP-C1(ClontechTM)。通过聚合酶不完全引物延伸法、使用如下引物和模板构建YFP-V7:来自pcDNA3.1AR-V7模板5'-GGT GCT GGA GCA GGTGCT GGA ATG GAAGTG CAG TTAGGGCTG和5'-GGAAAT AGG GTT TCC AAT GCT TCA GGGTCT GGT CATTTT GAG的AR-V7插入物;缺乏全长AR 5'-CAGCCC TAA CTG CAC TTC CAT TCC AGC ACC TGC TCC AG和5'-CTC AAA ATG ACC AGA CCC TGA AGC ATT GGAAAC CCT ATT TCC的pEYFPC1载体。
细胞培养、转染和荧光素酶测定
使PC3人PCa细胞(AATC)在补充5%活性炭处理的血清(CSS)的RPMI 1640培养基(InvitrogenTM)(含有5%CSS的RPMI 1640培养基)中血清饥饿5天,然后转染。为了进行荧光素酶测定,将PC3细胞接种入96-孔板(5000细胞/孔)中的具有5%CSS的RPMI 1640培养基24h,然后用50ng hAR或其它核受体质粒、50ng ARR3tk-荧光素酶和0.3μl/孔Trans IT20/20转染试剂(TT20,MirusTM)转染48h。然后用不同浓度的化合物和0.1nM R1881(在100%乙醇中)处理24h。分别用1nM地塞米松或孕酮刺激GR或PR活化。使用具有稳定转染的雌激素响应元件-荧光素酶报道分子的MCF-7细胞系测定ER-α转录活性,用1nM雌二醇刺激转录活性。使用60μl 1X被动性裂解缓冲液/孔(PromegaTM)进行细胞裂解。
将来自每个孔的20μl细胞裂解液与50μl荧光素酶测定试剂(PromegaTM)混合,并且用TECANTM M200pro平板读出器记录发光。按照相同方式使用剪接变体AR进行荧光素酶测定,但仅使用5ng pcDNA3.1AR-V7(以便限制高水平的AR-V7表达),且不使用R1881。R1-AD1和TALEN-改造的R1-D567细胞系如上所述(Nyquist,M.D.等人,(2013)TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer.Proc.Natl.Acad.Sci.U.S.A.110,17492-17497)。如上所述使用R1-AD1和R1-D567细胞进行测定,但仅转染ARR3tk-荧光素酶报道基因和10,000细胞/孔。
蛋白质印迹
用10%SDS-聚丙烯酰胺微型凝胶从荧光素酶测定(96-孔板)中分离细胞裂解液(40μl)。将蛋白质转入添加甲醇的PVDF膜并且用抗-AR441(小鼠,SigmaTM)单克隆初级抗体探测。还用显示等同载量的多克隆抗-肌动蛋白(家兔,SigmaTM)和多克隆抗-PARP/抗-裂解PARP(家兔,SigmaTM)探测印迹,以便测试细胞凋亡诱导。与化合物一起温育2天后,再用多克隆抗-FKBP5(家兔,SigmaTM)探测来自CWR-R1细胞的裂解液。
PSA测定
将维持在具有5%CSS的RPMI 1640培养基中的LNCaP细胞在96-孔(10,000细胞/孔)中的相同培养基中和化合物和1nM R1881的存在下温育2天。在温育期后,从每个孔中取出150μl培养基并且使用Cobas e411分析仪(Roche Applied ScienceTM)、根据制造商的说明书定量PSA水平。相同仪器用于分析体内分析过程中来自小鼠的血清PSA。
微阵列基因档案
使LNCaP细胞生长在如下4种条件下24h:1)DMSO,无R1881;2)DMSO与R1881(1nM);3)400nM化合物14449与R1881;和4)120nM恩杂鲁胺与R1881。化合物浓度遵循在荧光素酶报道分子测定法中测定的约IC50浓度。
一式三份重复每种条件。24h后,从12份样品中各自提取总细胞mRNA(4种条件,3次)并且从定制的Agilent微阵列中测定50,737个转录物的基因表达水平。
在全部样品中以分位数校准基因表达数据并且转化成log2等级。对条件3(化合物14449与R1881)与条件2(DMSO与R1881)和条件4(恩杂鲁胺与R1881)与条件2(DMSO与R1881)之间的每种转录物的表达水平进行两种-样品t检验。如果来自两种-样品t检验的p值小于0.05,则将基因视为差别表达的。Fisher确切检验和比值比用于评价不同组差别表达的基因之间的重叠。
共焦显微镜检查
将约40,000PC3细胞在置于12-孔板内在具有5%CSS的RPMI1640培养基中的无菌盖玻片上接种48h。使用TT20(3μl)将YFP-AR或YFP-V7质粒(100ng/孔)转染48h。然后用10nM R1881和25μM化合物将细胞处理6h。抽吸培养基后,用PBS将细胞洗涤1次,并且用4%低聚甲醛在4℃固定过夜,然后使用DAPI固定器(Vector Laboratories)在带电荷的盖玻片上固定。用Zen 2012软件控制的Zeiss LSM 780共焦旋转盘式显微镜取图像。分别使用508和388nm的激发波长使YFP和DAPI显影。
染色质免疫沉淀法(ChIP)
用单独的DMSO、DMSO+R1881或化合物+R1881将除去雄激素的LNCAP细胞处理24h。在室温下使用1%甲醛处理进行DNA-蛋白质交联10min,并且用125mM甘氨酸终止5min。用Thermo ScientificTM1/8-英寸超声处理探头和Sonic Dismembrator 550TM仪器将细胞裂解液(1X 107细胞/ml)进行超声处理,以便产生200-1000bp大小的DNA片段。使用5μg抗-AR-N20抗体(Santa Cruz BiotechnologyTM)或1μg家兔同种型控制IgG(Santa Cruz BiotechnologyTM)、应用EZ-ChIP〗染色质免疫沉淀试剂盒(MilliporeTM)对裂解液(3.3×106细胞等份)进行免疫沉淀。使用如下引物组,通过定量PCR(SYBR Green主要混合物,InvitrogenTM)对结合的DNA进行定量:PSA增强子,正向5'-ATGGAGAAAGTGGCTGTGGC和反向5'-TGCAGTTGGTGA GTG GTC AT;FKBP5增强子,正向5'-CCC CCCTAT TTT AAT CGG AGT AC和反向5'-TTT TGA AGAGCA CAG AAC ACC CT;GAPDH启动子,正向5'-TACTAGCGGTTTTACGGGCG和反向5'-TCGAACAGGAGC AGA GAG CGA。将定量的PCR结果呈现为使用控制IgG抗体的PCR扩增的折叠富集,并且给予总输入值(未沉淀的染色质)校准。将用于GAPDH启动子的引物用作缺乏任何雄激素响应元件的阴性对照。
AR-DBD蛋白质的纯化
将编码AR-DBD+铰链的质粒转入BL21(DE3)。将为表达生物素标记的AR-DBD+铰链设计的BL21细胞与Pan4AR-DBD+铰链(氨苄西林筛选)和生物素连接酶表达载体(pBir-Acm,氯霉素筛选)共转化。使单一克隆生长在补充了50μg/ml氨苄西林和35μg/ml氯霉素(如果适合)的2升LB培养基中至A600nm=0.6,然后在37℃下用0.1mM异丙基β-D-1-半乳糖硫吡喃糖苷诱导3h。在诱导步骤过程中同时给表达具有生物素化序列的AR-DBD的培养物补充0.150mM生物素。将细胞沉淀重新混悬于补充了10mM咪唑的~20ml 50mM Tris-HCl,pH 8.0、300mM NaCl、5%甘油(缓冲液A),并且与0.1mg/ml鸡卵白溶菌酶(SigmaTM)和0.1mM PMSF蛋白酶抑制剂一起在冰上温育30min。通过超声处理进行细胞裂解,然后在4℃以13,000X g离心30min。在4℃将上清液与2ml镍-琼脂糖珠(GE HealthcareTM)一起旋转1h,然后直接上Poly-Prep 10-ml重力色谱柱(Bio-RadTM)。用补充20mM咪唑的2X 4ml缓冲液A进行洗涤。用2ml缓冲液A洗脱包含250mM咪唑的500-μl级分中纯的蛋白质。
EMSA(凝胶转移)测定法和生物层干扰量度分析
使用纯化的AR-DBD和具有ARE 2序列的dsDNA进行电泳迁移位移试验(EMSA)。通过使乳腺互补寡核苷酸在H2O中退火形成ARE:上链,5'-TAC AAA TAG GTT CTT GG AGTACTTTA CTAGGCATG GAC AAT G;和下链,5'-CAT TGT CCA TGCCTAG TAA AGTACT CCA AGA ACC TAT TTG TA。给六聚化AREs的位置加下划线。使乱序的DNA从如下序列中退火:上链,5'-TAAAACGTGGTCCCTGGTACTGCCTTCGTGCCATTC GAT TTT;和下链,5'-AAA ATC GAA TGG CAC GAA GGC AGT ACC AGG GAC CAC GTT TTA。使蛋白质-DNA复合物在冰上在加样缓冲液(20mM Tris,pH 8、50mM NaCl、1mM EDTA、10μg/ml聚(dI-dC)、5mM MgCl2、200μl/ml BSA、5%甘油和1mM DTT)中温育30min,然后在6%天然-PAGE上的1X TBE,pH 8.0中电泳。使用SyberSafeTMDNA染色染料使蛋白质-DNA复合物显影。
在全部实验的至始至终,使用生物素化AR-DBD+铰链在具有5%DMSO的缓冲液A中进行使用ForteBio Octet RedTM仪器的生物层干扰量度分析。使DBD(0.1mg/ml)上链霉抗生物素传感器的200μl缓冲液中30min,然后用生物胞素(10μg/ml)封闭游离链霉抗生物素位点10min。然后用在相同缓冲液中的50μM化合物或单独的5%DMSO使加载DBD的传感器预平衡100s。通过使传感器运动入包含dsDNA(ARE,3μM)的补充了50μM化合物的孔120s监测DNA连接动力学。然后在缓冲液+化合物、但无DNA中解离,再经过120s。使生物胞素-封闭的对照传感器(无AR-DBD)接触相同的实验条件,并且从每个曲线上扣除与dsDNA的非特异性相互作用。
肿瘤生长评价和去势抗性LNCaP异种移植物的PSA
给称重25-31g的6-8-周龄裸鼠(Harlan Sprague-Dawley)的后背部位上皮下接种LNCaP细胞(BD Matrigel中106细胞,BD Biosciences)。每周测量肿瘤体积、体重和血清PSA水平。当血清PSA水平达到25ng/ml以上时,阉割小鼠。当PSA恢复至阉割前的水平时,将小鼠随机分入如下的3个治疗组:媒介物、10mg/kg恩杂鲁胺或100mg/kg化合物14449,并且通过每日2次腹膜内注射4周治疗。卡钳用于测量每个肿瘤的3个垂直轴以便计算肿瘤体积。另外,每周称重小鼠并且每日监测毒性症状,包括死亡、昏睡、失明和定向障碍。
实施例
生物信息学假说
1.AR DBD中潜在成药的结合位点的鉴定
在比较来自DBD二聚体-DNA复合物、DBD二聚体和DBD单体的全部鉴定的结合位点后,鉴定DBD单体中的潜在结合位点,如果化合物结合,则推定其能够打断DBD-DNA结合。该结合位点主要由来自一种α-螺旋的残基包括Arg568、Val564和Phe565和一些来自环路的极性残基Tyr576和Gln574组成。它是牵涉残基的区,所述残基构成了对DNA结合的关键相互作用,如形成范德瓦尔斯力的Arg568接触Val564和核苷酸(未显示)。
2.通过虚拟筛选法鉴定结合AR DBD的化合物
首先通过对接程序Glide对AR DBD亚单位中检测的结合位点筛选ZINC数据库。弃去具有对接评分大于(<-4)的总计462,588个化合物并且用分子量、电荷和环数过滤化合物,且将其余化合物(170,000)进行eHiTs对接。保留具有低于(<-3)的eHiTs评分的59,586个化合物,并且计算这些化合物的RMSD值。处理具有RMSD的8,953个化合物以便筛选潜在的虚拟命中数。通过对受体-配体相互作用进行视觉检查,选择100个化合物用于实验评价。
3.靶向AR DBD的AR抑制剂的体外鉴定
1.有效和多种AR抑制的鉴定
使用无破坏性eGFP测定法评价选择的化合物,所述无破坏性测定法对AR转录活性水平进行定性,并且鉴定属于不同化学类型的化合物(表1-系列1-6),且特别是4-(4-苯基噻唑-2-基)吗啉化合物(14228)显示出高效能的AR转录抑制。该化合物抑制AR转录并且以浓度依赖性方式抑制PSA水平,其中IC50s处于低纳摩尔范围(分别为0.274μM和0.188μM)。进一步确定了在3种细胞系AR-阳性LNCaP和MDV3100-抗性细胞系和AR-阴性PC3细胞系中抑制前列腺癌细胞生长的能力。显示该化合物强烈抑制AR-阳性前列腺癌细胞系,而对AR-阴性细胞无影响,表明该化合物的效能通过抑制AR进行(图3)。
4.不与AR中的其它已知结合位点发生相互作用的化合物14228
因为在AR中存在其它已知的结合位点,包括激素结合位点(HBS)、活化功能-2(AF2)位点、结合功能3(BF3)和N-末端活化功能-1(AF1)位点,所以我们使用另外的测定法以便排除可能与那些位点的结合。雄激素置换测定法证实,它不会置换雄激素以占据AR HBS,因此,它是非竞争性AR抑制剂。它不会置换通过针对AF2的荧光偏振肽置换测定法所显示的AF2结合位点中的肽。此外,生物层干扰量度(BLI)测定法显示,在C-末端配体结合结构域(LBD)中不存在结合,因此,它不是BF3结合剂。所有这些证据启示,所述化合物不结合LBD中的任意结合位点。然后,我们使用AF1测定法测定该化合物是否来自AF1反式激活的抑制。将CREB和Gal4质粒共转染入AR-阴性细胞系PC3,并且在处理后,无CREB和Gal4抑制,表明它不影响AF1(数据未显示)。
5.化合物结合AR DBD的直接证据
为了进一步如所推定的证实化合物结合AR DBD,我们对预测的结合位点进行了突变研究。基于我们的分子模型化,Val581、Phe582、Arg586、Glu591和Tyr593是可能具有氢键和与配套的疏水性相互作用的关键残基。因此,我们使这些残基突变成天冬氨酸并且使用这些突变体测试我们的化合物。在这些突变体中,当将Val581、Phe582和Arg586突变体与AR3TK-荧光素酶报道分子一起共转染入AR-阴性PC3细胞中时,所述突变体不能在荧光素酶试验中活化荧光素酶表达(图4),同时,在蛋白质印迹中未检测到AR表达(数据未显示)。所述化合物不抑制Glu591和Tyr593突变体(可以活化野生型AR),这启示该化合物结合AR DBD,而这两种残基对于结合亲和力而言是关键(图4)。
通过蛋白质消化试验进一步证实了化合物结合AR DBD。在化合物14288的存在下用胰蛋白酶无法消化AR DBD,而用媒介物或其它抗雄激素药如MDV3100可以消化(数据未显示)。
6.鉴定的化合物的特异性
因为DBD是核受体中高度保守的结构域时,所以靶向该结构域的化合物可能具有极差的选择性。为了排除非特异性的问题,我们测定我们的化合物在其它核受体如ER中抑制。在用ERE-荧光素酶报道基因转染的乳腺癌MCF7细胞中测试所鉴定的化合物,它们未显示对ER抑制的显著作用(数据未显示)。
7.对14228的结构修饰
因为起始命中的化合物14228显示有利的特性,所以购买或基于对接模型合成更多类似物(未显示)。使用14228的三部分组成苯环、噻唑和吗啉基团进行结构修饰(表1),且一些类似物如14370与亲代化合物相比显示改善的活性。
8.14368的部分AR激动剂属性的鉴定
为了防止对AR-LBD中突变产生的常规抗雄激素药的抗性,我们探查了AR的DBD片段上的交替成药结合位点,并且研发了一系列4-(4-苯基噻唑-2-基)吗啉类,它们能够通过破坏其与DNA的相互作用选择性地抑制受体活性。这类化学类型的一种衍生物即化合物14368在报道试验中显示对AR转录的极为有效的抑制和在R1881刺激后LNCaP细胞中表达AR靶基因PSA(相应的eGFP IC50和PSA IC50值分别为0.035μM和0.13μM)。然而,还观察到,这种抑制作用在高浓度所述化合物下可逆(图1A-B),这启示了激动剂作用。为了验证这些发现,我们使用相同的AR-调节的表达eGFP的LNCaP细胞、但不使用R1881刺激评价了所述化合物的作用。在没有雄激素的存在下,14368能够强有力地增强AR的转录活性(图1C)。重要地,所观察到的这种化合物的激动剂作用发生在携带T877A AR突变的LNCaP细胞中。为了验证这种作用与存在T877A突变相关,荧光素酶报道试验用于研究为AR阴性的PC3细胞中野生型AR和T877A-AR突变体的转录活性。14368按照剂量响应方式,在R1881的存在下抑制用野生型AR瞬时转染的PC3细胞中的AR转录,而在没有雄激素的存在下仅显示基础水平的转录活性(图1D)。相反,14368在高于0.4μM的浓度下,在R1881存在和不存在下诱导T877A AR突变体的转录活化(图1E)。此外,这种激动剂作用在包含受体的野生型形式的R1-AD1细胞中在高浓度14368下未观察到(Nyquist等人2013PNAS 110:17492-17497所述的方法中)(图1F)。这些观察结果共同证实14368作为针对野生型AR的拮抗剂起作用,而在较高浓度下,在T877A AR突变存在下,它变成激动剂。
9.4-(4-苯基噻唑-2-基)吗啉类的部分激动作用的消除
为了消除观察到的结合人AR的LBD和得到的对AR的部分激动剂作用,我们尝试用疏水性较低的杂环替代所研究的4-(4-苯基噻唑-2-基)吗啉类中的苯基碎片。生成了大量包含不同芳族(14291)、脂族(14403、14406)和杂环(14404、14435、14436、14439)的替代了苯碎片的化合物(参见表2)。这些化学品是由公司Life Chemicals(www.lifechemicals.com)和Enamine(www.enamine.net)定制合成的。其纯度和特性分别通过LC-MS和1H NMR证实。
使用荧光偏振测定法(PolarScreenTM,Life Technologies)在不同浓度下测定上述化学品置换来自重组野生型LBD的ABS位点的DHT的能力。按照浓度依赖性方式检测到14368结合ABS,其中在~30μM建立了相应的IC50(表2,图2)。用疏水性较低的杂环和庞大的脂族环替代苯环可以显著地降低化合物的DHT置换能力。尽管衍生物的一些(14291、14403和14406)失去其抗-AR活性,但是其它的化合物例如14435、14436、14439和14404维持了对AR的突变的T877A形式的良好效能,同时未显示出不期望的部分激动剂作用。
表2:4-(4-(3-氟-2-甲氧基苯基)噻唑-2-基)吗啉衍生物的结构和活性
10.AR DBD表面上潜在成药结合位点的鉴定
结合两个六聚化半位点AREs(PDB代码:1R4I)的大鼠AR DBD二聚体是迄今为止可得到的受体的DBD区的唯一晶体结构。因为大鼠和人AR DBDs的序列相同,所以将1R4I结构用作构建人AR DBD的同源性模型的模板。有关AR DBD二聚体-ARE复合物“热点”根据Molecular Operating Environment(MOE)2011包内的Site Finder模块预测。将AR DBD的P-框区下的空穴视为可能打断AR DBD-ARE复合物的小分子结合的潜在位点(图5)。该空穴主要被识别螺旋的残基Ser579、Val582、Phe583和Arg586和属于杠杆臂环路的极性残基Gln592和Tyr594和来自另一α-螺旋的残基Pro613和Arg616与环路残基Arg609和Lys610包被。该位点以有溶解能力地暴露于特定残基,预测这些残基在锚定小分子的可能结合方面起关键作用。因此,该位点周围的极性残基Ser579、Gln592和Try594可以被表征为可利用于氢键,而该位点核心中的Phe583可以提供额外的与潜在结合剂的疏水性相互作用。
11.化学
全部试剂和溶剂购自商品供应商并且无需进一步纯化使用,另有描述的除外。通过预涂布的硅胶F254板(Sigma-Aldrich)上的薄层色谱法(TLC)与UV指示剂、使用乙酸乙酯/己烷(1:2v/v)监测反应。收率是纯化产物的收率且未优化。通过LCMS分析测定新合成的化合物的纯度。使用具有400MHz操作频率的Varian GEMINI 2000NMR分光光度计系统显示质子核共振(1H NMR)光谱。以ppm给出化学位移δ并且使用如下缩写:单峰(s)、双峰(d)、三重峰(t)、四重峰(q)、多重峰(m)和宽单峰(br s)。用Agilent 1100 LC系统采集全部LC/MS数据。将化合物溶液注入以正和负模式操作的电离源,其中流动相为乙腈/水/甲酸(50:50:0.1%v/v),1.0mL/min。在外部对质量范围m/z100-m/z 650校准仪器。
方案1.用于合成杂芳基衍生物14449(25)、14408(27)、14404(28)、14451(31)、14402(32)、14448(34)、14450(36)、14464(38)、14468(40)和14447(41)的方案。
(A)
(B)
(C)
试剂和条件:(A)IPA,100℃,17-38%;(B)THF,H2O,K2CO3,PdCl2(PPh2C6H4SO3Na-m)2,80℃,54-73%;(C)甲苯,PdAc2(PPh3)2,110℃,1062%。
化合物27的合成方法(方案1(A))。将10mmol硫脲(a)和10mmol卤代酮(b)的混合物溶于10mL异丙醇。将形成的溶液回流4h,真空除去有机溶剂。用20mL盐水处理残余物,用30mL乙酸乙酯萃取(2次)。用硫酸钠干燥合并的有机层,过滤,蒸发。通过制备型HPLC(洗脱液EtOAc/己烷=1/1)纯化得到的固体,得到终产物,为固体。
4-(4-(4,5-二甲基噻吩-3-基)噻唑-2-基)吗啉(27)。1H NMR(DMSO-d6,400MHz):2.24(3H,s),2.36(3H,s),3.41-3.43(4H,m),3.73-3.76(4H,m),6.76(1H,s),7.33(1H,MS(ESI):m/z(M+H)+281.1。收率:38%;LCMS纯度100%。
化合物28的合成方法(方案1(B))。将9.1mmol溴噻唑(bromthiazole)(方案1(B),c)、11.8mmol相应的硼酸、22.5mmol碳酸钾和0.1mmol催化剂PdCl2(PPh2C6H4SO3Na-m)2的混合物溶于100mL四氢呋喃和16mL水。然后将该反应混合物回流20h。
冷却后,用100mL乙酸乙酯萃取。分离有机层,用硫酸钠干燥,过滤,蒸发。通过制备型HPLC(洗脱液EtOAc/己烷=1/1)纯化得到的残余物,得到终产物,为固体。
4-(4-(噻吩-3-基)噻唑-2-基)吗啉(28)。1H NMR(DMSO-d6,400MHz):3.41-3.43(4H,m),3.72-3.74(4H,m),7.14(1H,s),7.49-7.55(2H,m),7.72-7.73(1H,m)。MS(ESI):m/z(M+H)+253.1。收率:54%;LCMS纯度95%。
用于合成化合物14449(25),14408(27)、14404(28)、14451(31)、14402(32)、14448(34)、14450(36)、14464(38),14468(40)和14447(41)的通用方法(方案1(C))。将4mmol溴噻唑(c)、12mmol相应的咪唑(d)、0.1mmol催化剂PdAc2(PPh3)和50mL甲苯的混合物回流24h。冷却后,真空除去有机溶剂。通过制备型HPLC(洗脱液EtOAc/己烷=1/1)纯化得到的残余物,得到终产物,为固体。再通过反相HPLC(洗脱液乙腈/水)纯化化合物,其中乙腈含量为20-80%。
4-4(4,5-二溴-1H-咪唑-2-基)吗啉(25)。1H NMR(DMSO-d6,400MHz):3.39-3.42(4H,m),3.70-3.73(4H,m),7.14(1H,s),7.80(1H,s)。MS(ESI):m/z(M+H)+395.0。收率:51%;LCMS纯度98%。M.p.116-118℃。
4-(4-(5-溴-4-氯-1H-咪唑-1-基)噻唑-2-基)吗啉(31)。1H NMR(DMSO-d6,400MHz):3.41-3.43(4H,m),3.71-3.73(4H,m),7.15(1H,s),8.16(1H,s)。MS(ESI):m/z(M+H)+351.0。收率:14%;LCMS纯度96%。
4-(4-(4,5-二氯-1H-咪唑-1-基)噻唑-2-基)吗啉(32)。1H NMR(DMSO-d6,400MHz):3.41-3.45(4H,m),3.71-3.73(4H,m),7.15(1H,s),8.14(1H,s)。MS(ESI):m/z(M+H)+305.1。收率:48%;LCMS纯度99%。
4-(4-(4-氯-1H-咪唑-1-基)噻唑-2-基)吗啉(34)。1H NMR(DMSO-d6,400MHz):3.42-3.44(4H,m),3.71-3.73(4H,m),6.97(1H,s),7.83(1H,s),8.17(1H,s)。MS(ESI):m/z(M+H)+271.1。收率:62%;LCMS纯度99%。
4-(4-(4-溴-5-氯-1H-咪唑-1-基)噻唑-2-基)吗啉(36)。1H NMR(DMSO-d6,400MHz):3.41-3.43(4H,t),3.70-3.72(4H,m),7.14(1H,m),8.15(1H,s)。MS(ESI):m/z(M+H)+351.0。收率:27%;LCMS纯度96%。
4-(4-(4,5-二碘-1H-咪唑-1-基)噻唑-2-基)吗啉(38)。1H NMR(DMSO-d6,400MHz):3.41-3.42(4H,m),3.71-3.73(4H,m),7.10(1H,s),8.10(1H,s)。MS(ESI):m/z(M+H)+488.9。收率:12%;LCMS纯度96%。
4-(4-(4-氯-5-碘-1H-咪唑-1-基)噻唑-2-基)吗啉(40)1H NMR(DMSO-d6,400MHz):3.41-3.42(4H,m),3.70-3.71(4H,m),7.14(1H,s),8.14(1H,s)。MS(ESI):m/z(M+H)+397.5。收率:10%;LCMS纯度96%。
4-(4-(4-溴-1H-咪唑-1-基)噻唑-2-基)吗啉(41)。1H NMR(DMSO-d,6400MHz):3.42-3.43(4H,m),3.71-3.74(4H,m),6.98(1H,s),7.88(1H,s),8.18(1H,s)。MS(ESI):m/z(M+H)+316.1。收率:53%;LCMS纯度99%。
用于其余化合物(4-4-(3,4-二氟-2-甲氧基苯基)噻唑-2-基)吗啉(26)、2-氟-6-(2-吗啉代4-基)苯酚(29)、4-(4-(4-氟-2-甲氧基苯基)噻唑-2-基)吗啉(30)、2,3-二氟-6-(2-吗啉代4-基)苯酚(33)、5-氟-2-(2-吗啉代4-基)苯酚(35)、4-(5-甲基-4-苯基噻唑-2-基)吗啉(37)、4-(4-吡啶-2-基)噻唑-2-基)吗啉(39)、4-(4-(吡啶-4-基)噻唑-2-基)吗啉(42)、3-氟-4-甲氧基-5-(2-吗啉代4-基)苯酚(43)和4-(4-(3-氟-2-甲磺酰基-苯基)-噻唑-2-基)吗啉(44)和化合物(72-84)的合成方法遵循上文。
本文所述的化合物可以如本文所述使用本文所述的改进方法或通过本领域技术人员公知的方法合成。
附加的参考文献
Lack,N.A.,P.Axerio-Cilies等人,(2011)。Journal of Medicinal Chemistry 54(24):8563-73。
Tavassoli,P.,R.Snoek等人,(2007)。Prostate 67(4):416-426。
更详细地举出如下实施例(即12-17),Dalal K.等人,“Selectively Targeting the DNA-binding Domain of the Androgen Receptor as a Prospective Therapy for Prostate Cancer”THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL.289、NO.38,pp.26417-26429,2014年9月19日(在线公布,2014年8月1日)。
12.荧光素酶报道试验
使用PC3细胞中由基于ARR3tk前列腺碱性蛋白的启动子驱动的荧光素酶报道试验(Snoek,R.等人,(1998)Differential transactivation by the androgen receptor in prostate cancer cells.Prostate 36,256-263),预测结合AR-DBD的两种化合物显示对瞬时表达的全长人AR的剂量依赖性抑制作用(数据未显示,14228和14449,相应地,IC50=2.36和0.340μM),不影响AR蛋白质表达(数据未显示)。近期报道14337(扑蛲灵)通过靶向DBD抑制全长和剪接变体AR形式(Lim,M.等人,(2014)Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium.ACS Chem.Biol.9,692-702),并且还可以抑制AR转录活性(数据未显示,IC50=0.194μM)。对照实验证实14449可以将AR抑制至与恩杂鲁胺相差无几的水平(数据未显示,IC50=0.314μM)。针对PARP的蛋白质印迹证实化合物处理不影响总PARP水平,也不产生任何PARP裂解产物,除了14337(数据未显示)之外。这些结果表明扑蛲灵(14337)强有力地诱导细胞凋亡,而DBD抑制剂(14228/14449)显示几乎物毒性或无毒性。
为了验证AR-DBD结合剂的作用位点,我们在预测与先导化合物发生相互作用的残基上导入了点突变。在相关核受体中存在氨基酸差异(数据未显示)的区中鉴定的两个位置(Tyr-594和Gln-592)可以具有天冬氨酸取代,但没有消除全长全长AR活性。而Y594D和Q592D突变体可以被恩杂鲁胺抑制,荧光素酶表达不受14228和唯一高浓度(≥25μM)14449(数据未显示)影响。相反,扑蛲灵强烈地抑制两种AR突变体(数据未显示),这启示该化合物衔接表面暴露的袋中非Tyr-594/Gln-592的残基或结合DBD表面上的不同位置。蛋白质印迹分析证实突变体AR蛋白质的表达未因药物抑制作用而改变(数据未显示)。在这些位置上导入酸性残基可以防止对支持化合物结合必不可少的疏水性相互作用。
除将带电荷的氨基酸导入DBD外,我们还测试了Y594A和Q592A突变体,它们两者可以被14228/14449抑制,但与野生型AR(数据未显示)相比具有明显较高的IC50值(数据未显示,~3-6μM)。可能的情况是,除去Gln或Tyr侧链在袋中生成了额外的空间,以使化合物进入,但抑制AR活性的能力降低。得到的IC50值增加进一步支持Tyr-594和Gln-592残基对化合物结合的重要性,在其侧链被除去后,这种结合受损。为了确定我们的化合物是否与相关核受体的DBDs发生交叉反应,我们使用全长ER、GR和PR进行测定(图6)。荧光素酶构建体包含具有用于AR/GR/PR的ARR3tk的相应核受体的适合的响应区和用于ER的雌激素效应元件。14228/14449显示在高于5μM的浓度下抑制ER转录活性(图6B和C,黑色条),但在与抑制AR(白色条)相比时有效性降低几倍。全部3种测试化合物对全长GR和PR的转录活性完全无效,甚至在以25μM浓度施用时也是如此(图6,分别为深灰色和浅灰色条)。值得注意地,恩杂鲁胺显示对全长ER的显著的交叉反应性,达到与对AR相差无几的抑制水平(图6,黑色条)。这些数据启示所研发的化合物具有对AR的有效抑制作用、结合ARDBD上预期的靶位点并且显示几乎无交叉反应性或无交叉反应性。
13.DBD-相互作用化合物减量调节LNCaP细胞中雄激素应答基因的表达
为了评价14228/14449阻断天然存在的AR-调节基因的转录能力,同时用R1881和化合物处理LNCaP细胞,然后测定分泌的PSA(图7A)。结果显示使用14228/14449和恩杂鲁胺的剂量依赖性抑制作用,其中在亚微摩尔浓度下建立了全部相应的IC50值。
使用化学-基因组方法(Bredel,M.和Jacoby,E.(2004)Chemogenomics:an emerging strategy for rapid target and drug discovery.Nat.Rev.Genet.5,262-275),应用先导抑制剂14449对基因表达进行更深入的分析。为了测定对雄激素-或基因毒素-应答基因表达的任何作用,用14449(400nM)或恩杂鲁胺(120nM)在R1881的存在下处理LNCaP细胞,以便使用AgilentTM基因表达微阵列比较转录应答(图7B)。几种众所周知的AR靶基因(Nelson,P.S.等人,(2002)The program of androgen-responsive genes in neoplastic prostate epithelium.Proc.Natl.Acad.Sci.U.S.A.99,11890-11895;和Magee,J.A.等人,(2006)Direct,androgen receptor-mediated regulation of the FKBP5 gene via a distal enhancer element.Endocrinology 147,590-598),包括KLK3(PSA)、KLK2、TMPRSS2和FKBP5在R1881的存在下增加基因表达(比较DMSO+R1881与DMSO-R1881),其中倍数改变分别为2.42、3.60、2.25和1.61。在用14449+R1881处理后,KLK3、KLK2和TMPRSS的基因表达与仅用R1881处理相比均显著地降低,分别有0.82、0.66和0.69的倍数改变。使用14449对这些AR靶基因表达的降低与使用恩杂鲁胺相差无几,倍数改变为0.71、0.64和0.62。使用14449的FKBP5表达减少(倍数改变0.81);然而,0.08的p值不满足基于两种-样品t检验的0.05截止值,可能归因于样品尺寸较小。化合物和恩杂鲁胺显示对非雄激素调节基因β-肌动蛋白(ACTB)表达无影响。为了鉴定其它减量调节的基因,将如下标准应用于全部用AgilentTM微阵列测定的50,737个转录物:1)基于两种样品t检验的p值<0.05;和2)倍数改变≤0.85。14449减量调节总计354个基因,其中112个也被恩杂鲁胺减量调节。两种化合物之间的减量调节的基因重叠是显著性的,其中Fisher确切检验的p值小于2.20E-16,且比值比为45.86。此外,将14449和/或恩杂鲁胺减量调节的基因清单与显示预先在LNCaP细胞中被雄激素增量调节的总计86个基因比较(Nelson,P.S.等人,(2002)The program of androgen-responsive genes in neoplastic prostate epithelium.Proc.Natl.Acad.Sci.U.S.A.99,11890-11895)。14449的354个减量调节基因的12个被雄激素增量调节(显著重叠:p值=2.73E-08,比值比=9.55),且恩杂鲁胺的314个减量调节的基因的15个被雄激素增量调节(显著重叠:p值=3.34E-12,比值比=14.27)。10个基因中有基因KLK2、KLK3(PSA)和TMPRSS2,它们被雄激素增量调节,但被14449和恩杂鲁胺减量调节。因14449或恩杂鲁胺处理产生的两组相互排斥的减量调节的基因显示了每种化合物抑制AR信号传导的作用的明显不同的机制。最终,为了鉴定来自两种化学品的任何潜在的基因毒性作用,将14449或恩杂鲁胺增量调节的基因与31个基因清单比较,其表达已经预先被证实在基因毒素的存在下增加(Ellinger-Ziegelbauer,H.等人,(2009)Characterization and interlaboratory comparison of a gene expression signature for differentiating genotoxic mechanisms.Toxicol.Sci.110,341-352)。该组基因毒素-应答基因包括大量p53靶基因牵涉细胞凋亡、DNA修复、DNA损伤应答和应激反应的其它基因。基因毒素-应答基因中无一显示因14449导致的任何显著性改变。未受影响的3种已知有代表性的基因包括CASP1(半胱氨酸天冬氨酸蛋白酶1,细胞凋亡,倍数改变=1.00,p值=0.73)、XPC(着色性干皮病互补基因C,DNA修复,倍数改变=1.03,p值=0.59)和ATF3(转录激活因子3,应激反应,倍数改变0.99,p值=0.89)。结果共同证实所研发的AR DBD抑制剂可以显著地减量调节已知AR-调节基因的表达,从而达到与使用恩杂鲁胺相差无几的水平,其中无诱导的细胞毒性。
14.DBD-相互作用化合物抑制AR剪接变体的转录活性
因为大部分AR剪接变体保留DBD结构域,所以我们测试了对AR-V7转录活性抑制。使用相同的荧光素酶报道基因测定法,使用递增浓度的14228/14449或扑蛲灵降低瞬时表达的AR-V7的活性,不改变AR-V7表达(数据未显示)。
使用恩杂鲁胺的对照实验证实对AR-V7活性无作用(数据未显示),并且与无LBD存在下来自这种变体的结果一致。值得注意地,当与全长受体的抑制作用比较时,所述化合物无法实现完全抑制且对AR-V7的转录活性的有效性较低(IC50=4-8μM)。我们有充分理由认为,全长和剪接变体ARs的DBDs可以共有相似的蛋白质结构。因此,我们将Y594D突变导入AR-V7编码序列,以便测定该突变是否可以消除药物抑制作用。AR-V7Y594D的转录活性可以不受14228/14449抑制(数据未显示),这启示所述化合物的结合位置在所有AR形式上是类似的。与具有该突变的全长AR一样,AR-V7Y594D仍然可以被扑蛲灵强烈地抑制(数据未显示)。
瞬时AR-V7表达可能无法反映出细胞中的生理蛋白质浓度。为了研究我们的化合物对内源性表达水平的作用,我们使用一对表达全长AR(R1-AD1)或AR v567es变体(R1-D567)的同基因细胞系。R1-D567细胞通过TALEN-介导的AR外显子5-7缺失来源于R1-AD1细胞系,反映出在患者来源的LuCaP 86.2异种移植物组织中发现的AR基因重排(Nyquist,M.D.等人,(2013)TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in protaste cancer.Proc.Natl.Acad.Sci.U.S.A.110,17492-17497)。在将ARR3tk-荧光素酶质粒转染入这些细胞系后,14228/14449可以以递增浓度抑制野生型和AR v567es转录活性(数据未显示)。蛋白质印迹证实所述化合物对AR的任一形式的蛋白质表达无作用(数据未显示)。我们还对天然存在的AR-调节的FK506-结合蛋白5(FKBP5)进行了蛋白质印迹。在用14228/14449处理后,FKBP5蛋白质表达在R1-AD1和R1-D567细胞中减少,而仅用恩杂鲁胺处理影响表达全长AR的R1-AD1细胞(数据未显示)。这些结果与R1-D567细胞中v567es的siRNA敲除后观察到的FKBP5mRNA水平下降一致(Nyquist,M.D.等人,(2013)TALEN-engineered AR gene rearrangements reveal endocrine uncoupling of androgen receptor in prostate cancer.Proc.Natl.Acad.Sci.U.S.A.110,17492-17497)。
15.DBD-相互作用化合物不阻碍AR核转位
认为恩杂鲁胺和其它ARLBD抑制剂阻断AR的核定位,由此防止其启动转录(Ferraldeschi,R.等人,(2013)Abiraterone and novel antiandrogens:overcoming castration resistance in prostate cancer.Annu.Rev.Med.64,1-13;和Rathkopf,D.和Scher,H.I.(2013)Androgen receptor antagonists incastration-resistant prostate cancer.Cancer J.19,43-49)。与这种常规的抗雄激素机制相反,我们预期DBD-相互作用化合物可以发挥其对核AR的作用。为了测试这种理念,我们用编码YFP-标记的全长AR(黄色荧光蛋白,YFP-AR-van Royen,M.E.等人,(2012)Stepwise androgen receptor dimerization.J.Cell Sci.125,1970-1979)和剪接变体AR-V7(YFP-V7)的质粒转染PC3细胞。YFP-AR和YFP-V7能够驱动荧光素酶表达,并且可以被14228/14449抑制,表明YFP标记不影响AR转录活性或化合物抑制(数据未显示)。在用R1881和恩杂鲁胺处理时,共焦显微镜检查影像揭示出YFP-AR在细胞溶胶中与对照实验相比相差无几的水平(数据未显示)。相反,14228/14449无法防止R1881-刺激的YFP-AR的核定位,其中未观察到细胞溶胶中的荧光信号(数据未显示)。对照实验显示恩杂鲁胺或14228/14449在无R1881存在下不会刺激任何核定位(数据未显示)。YFP-V7在所有条件下、甚至在没有R1881的存在下均完全定位于核中(数据未显示),这与剪接变体自发地进行核转位的已知特性一致(Watson,P.A.等人,(2010)Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor.Proc.Natl.Acad.Sci.U.S.A.107,16759-16765)。这些结果共同启示,我们的化合物影响细胞核门AR及其剪接变体的活性,与直接影响DBD功能一致。
16.化合物在染色质水平和体外减少AR的DNA结合
由于预期所研发的AR DBD抑制剂结合AR-DBD上接近的蛋白质-DNA界面,所以所述化合物应当影响AR结合雄激素调节的基因的增强子(具有AREs)。在对来自用R1881和化合物处理的LNCaP细胞的染色质进行ChIP分析(AR-N20抗体)后,14428和14449与用单独的R1881处理相比减少了PSA和FKBP5增强子的AR折叠(数据未显示)。对照实验揭示出在恩杂鲁胺处理后或在使用GAPDH启动子阴性对照的任何条件下,在无R1881的存在下,无增强子破坏。
由于我们的化合物无法阻断核转位(数据未显示),所以ChIP结果启示14228/14449阻断AR与核中雄激素效应元件发生相互作用。为了直接探测DNA相互作用,我们研究了重组AR-DBD与包含两种六聚化AREs的寡核苷酸的结合。将纯化的人AR-DBD和铰链区(残基558-689,AR-DBD+铰链)与ARE 42-bp双链DNAs(dsDNA)一起温育并且通过天然-PAGE分析。该蛋白质能够在ARE与乱序的对照品之间区分(数据未显示),但14228/14449不能防止蛋白质-DNA复合物形成(数据未显示)。我们推断凝胶替换可能不足以检测到DNA结合中小的、但明显的改变,因为丙烯酰胺基质可以使疏水性化合物从蛋白质表面解离。为了防止这种限制,我们使用连接至链霉抗生物素包被的传感器的生物素化AR-DBD以用于生物层干扰量度分析。使生物素化DBD在DMSO或化合物的存在下暴露于ARE寡核苷酸。所观察到的结合动力学揭示出在14228/14449的存在下,与恩杂鲁胺和DMSO对照品相比,dsDNA结合的速率明显较缓慢,不过,解离保持相对不变(数据未显示)。使用具有Y594D突变的生物素化AR-DBD进行的相同实验直接地揭示出并且证实尽管与其它受体具有高度序列保守性,但是对转录活性具有特异性抑制。来自ER、GR和PR的相关DBD结构域可以与AR-DBD具有足够的结构差异,使得表面暴露的袋改变或不存在。值得注意地,AR的Gln-592不是与任意其它相关核受体一样保守的。该残基与Tyr-594可以促成独特的形状和DBD上表面暴露的袋的化学,且由此在突变时抵抗药物抑制。
可以预期通过诱变改变表面暴露的或埋藏的袋的特性。例如,T877A的单一氨基酸取代将AR交联剂转化成激动剂(Taplin,M.E.等人,(1995)Mutation of the androgen receptor gene in metastatic androgen-independent prostate cancer.N.Engl.J.Med.332,1393-1398),且这种转化可以用与药物复合的AR-LBD的晶体结构进行理论说明(Lallous,N.等人,(2013)Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer.Int.J.Mol.Sci.14,12496-12519)。此处,AR的Tyr-594和Gln-592上的天冬氨酸或丙氨酸残基取代对两种化合物抑制AR转录活性的能力具有显著影响,从而提供了对DBD结构域的作用的令人瞩目的证据。不排除化合物与AR其它结构域之间的可能的相互作用,但事实上,剪接变体的转录活性可能因对DBD的一些偏好的争议而受到强烈影响(数据未显示)。值得注意地,14449在≥25μM浓度下展示出对Y594D和Q592D突变体的抑制作用(数据未显示),这启示任一突变都可以使化合物的结合平衡移动,或实际上,14449能够衔接作为第二靶标的LBD或NTD结构域。
在与全长AR比较时,14228/14449的抑制作用对于剪接变体较弱。可以达到对于瞬时表达的V7的70-90%抑制率的最大值(数据未显示)和对于内源性产生的V567es的50-70%(数据未显示)。这些抑制水平与EPI-001的抑制水平类似,当在浓度25μm下对与ARR3tk-荧光素酶报道基因共转染的PC3细胞测试时,可以达到对AR(1-653)截短的缺乏LBD的突变体~80%的抑制率(Andersen,R.J.等人,(2010)Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domainof the androgen receptor.Cancer Cell 17,535-546)。
我们证实在已知抗雄激素药(恩杂鲁胺)和14228/14449的存在下,YFP-AR的核定位特性之间存在显著性差异(数据未显示)。尽管未受阻碍的核定位不会严格地排除14228/14449对LBD的作用,但是清楚地说明不同于恩杂鲁胺活性的机制,这促进了YFP-AR在细胞溶胶中的大量保留,可能通过替换R1881和改变AR蛋白质结构来进行(Watson,P.A.等人,(2010)Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor.Proc.Natl.Acad.Sci.U.S.A.107,16759-16765;和Tran,C.等人,(2009)Development of a second-generation antiandrogen for treatment of advanced prostate cancer.Science 324,787-790)。还启示14228/14449作用必须发生在核内,基需要破坏蛋白质-DNA相互作用。YFP-V7在药物的存在下或无药物存在下完全定位于核(数据未显示)这一事实也与预先的报道一致,表明恩杂鲁胺不能导致AR-V7再进入细胞溶胶(Watson,P.A.等人,(2010)Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor.Proc.Natl.Acad.Sci.U.S.A.107,16759-16765)。
所观察到的核定位动力学与化合物影响AR结合染色质或改变dsDNA以纯化的AR-DBD结合的能力一致(数据未显示)。14228/14449可能无法完全消除DNA结合,但是可以按照这种方式相当地弱化或调节这种结合,以便防止核AR的专利激活。澄清14228/14449干扰AR结合雄激素效应元件的确切模式是重要的研究领域。
17.体内异种移植试验
最终,我们证实化合物14449在体内具有有利的治疗特征(图8)。除对动物没有可观察到的毒性作用外,还抑制肿瘤体积和PSA表达至与恩杂鲁胺治疗相差无几的水平。因此,靶向AR的DNA结合在体内可以与用恩杂鲁胺防止核转位(Tran,C.等人,(2009)Development of a second-generation antiandrogen for treatment of advanced prostate cancer.Science 324,787-790)或通过用EPI-001阻断AR-NTD上的辅因子募集(Andersen,R.J.等人,(2010)Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor.Cancer Cell 17,535-546)同样有效。14449影响来自各种其它细胞系(雄激素敏感性的和雄激素依赖性的)的肿瘤异种移植物的能力面前正在进行中。
近期,据报道化合物恩波吡维铵(14337)抑制全长/剪接ARs的转录活性(Lim,M.等人,(2014)Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium.ACS Chem.Biol.9,692-702)。AR-DBD-DNA界面的模型化(蛋白质数据库代码1R4I)用于理论说明扑蛲灵与本文提出的相同表面暴露的袋发生相互作用,但在Lys-610至Pro-613的保守区中能够解释该化合物与ER和GR的交叉反应性(Lim,M.等人,(2014)Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium.ACS Chem.Biol.9,692-702)。未给出与这种袋发生喜欢着有的直接证据,但用LexA蛋白质替代全长AR上的DBD防止了药物抑制作用,这启示扑蛲灵结合在DBD上的某处(Lim,M.等人,(2014)Ligand-independent and tissue-selective androgen receptor inhibition by pyrvinium.ACS Chem.Biol.9,692-702)。在本文中,我们揭示出扑蛲灵的一般毒性(数据未显示)且不能抑制具有Q592D和Y594D突变的AR。尽管扑蛲灵强烈地抑制雄激素信号传导途径,但是其与核受体的交叉反应性和通过以纳摩尔浓度结合酪蛋白激酶家族成员破坏Wnt/β-cat信号传导(Thorne,C.A.等人,(2010)Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α.Nat.Chem.Biol.6,829-836)使得特异性AR抑制的适合性不清楚,最低限度至研发了该化合物的较少的偶然衍生物为止。
具有对AR剪接变体的活性的特异性AR-DBD抑制剂的鉴定对于治疗恩杂鲁胺抗性或或AR-变体驱动的去势抗性Pca而言具有极佳的潜能。
更具体地举出如下实施例(即18),Li,H.等人,(2014)Discovery of small-molecule inhibitors selectively targeting the DNA-binding domain of the human androgen receptor.J.Med.Chem.10.1021/jm500802j。
18.14449的AR I抑制活性、PSA抑制和抗增殖活性
具有4,5-二溴咪唑的合成类似物25(即14449)与化合物6(即14228)相比展示出2-倍的活性改善(eGFP IC50=0.12±0.01μM;PSA IC50=0.17μM,图9A和B),达到最新FDA批准的PCa药物恩杂鲁胺的活性水平(eGFP IC50=0.11±0.01μM;PSA IC50=0.12μM)。此外,化合物25(即14449)和其它包含卤素的类似物(Estebanez-Perpina,E.等人,A surface on the androgen receptor that allosterically regulates coactivator binding.Proc.Natl.Acad.Sci.U.S.A.2007,104,16074-16079;和Li,H.等人Identification of novel androgen receptor antagonists using structure-and ligand-based methods.J.Chem.Inf.Model.2013,53,123-130.)在介质中是完全稳定的。由于这种对AR转录和PSA表达的富有希望的抑制活性,所以我们进一步评价了25抑制AR-依赖性PCa细胞在AR-阳性LNCaP系统中生长的能力。在AR的激素活化后(0.1nM R1881),化合物25(即14449)引起细胞生长的浓度依赖性抑制。当对新近研发的恩杂鲁胺抗性细胞系MR49F评价25(即14449)时,实现了对细胞生长抑制的类似效能(Kuruma,H.等人,A novel antiandrogen,Compound 30,suppresses castration resistant and MDV3100-resistant prostate cancer growth in vitro and in vivo.Mol.Cancer Ther.2013,12,567-576)。重要地,化合物25(即14449)在抑制AR-阴性PC3细胞增殖中无效,这支持了其通过与AR的特异性相互作用起效、而非通过一般毒性的方式起效的机制(图9C)。
产物,对于预测的结合位点残基的定点诱变研究显示化合物14228和14449直接结合该位点。将野生型hAR和hAR突变体质粒(S579D、V582D、F583D、R586D、Q592D,Y594D和K610D)ARR3tk荧光素酶报道基因一起转染入PC3细胞,并且基于发光测定测试化合物14228、14449和恩杂鲁胺在10μM下的转录活性。经证实化合物14228和14449以浓度依赖性方式抑制AR-V7转录。将野生型hAR/ARV7质粒与ARR3tk-荧光素酶报道基因共转染入PC3细胞并且用化合物14228、14449和恩杂鲁胺处理。
尽管在此公开了本发明的各种实施方案,但是可以根据本领域技术人员的公知常识在本发明范围内进行许多改编和改动。这样的改动包括用已知等价物替代本发明的任一方面,以便以基本上相同的方式获得相同的结果。数字范围包括定义该范围的数字。措词“包含”在本文中作为开放式术语使用,基本上等价于短语“包括、但不限于”,并且措词“包括”具有相应的含义。如本文所用,单数形式“一个(a)”,“一种(an)”和“所述(the)”包括复数指代,除非上下文有清楚相反指示。因此,例如,提到“一个事情(a thing)”包括多于一个这样的事情。本文中对参考文献的引用不是承认这些参考文献是针对本发明实施方案的现有技术。特别地和分别地将本说明书中引述的任何优先权对比文件和所有的出版物包括、但不限于专利和专利申请引入本文参考,如同指示将每篇出版物特别地和分别地引入本文参考一样,不过,没有完整地举出。本发明包括如上文所述的所有实施方案和变化形式,并且参照了实施例和附图。
- 还没有人留言评论。精彩留言会获得点赞!
- 作为雄激素受体拮抗剂的n-[4-(喹啉-4-基氧基)环己基(甲基)](杂)芳基甲酰胺、其制法 ...的制作方法
- 一种用于雄激素受体转录后修饰位点预测的体外迷你报告基因及应用
- 膜雄激素受体(mAR)调节剂的检测方法
- 作为尿压素Ⅱ受体拮抗剂的4-(哌啶基-吡咯烷基-烷基-脲基)-喹啉类化合物的制作方法
- 作为抗雄激素剂的17α
- 亚甲基桥连的选择性雄激素受体调节剂及其应用方法
- 可用于治疗雄激素受体相关疾病的吲哚类化合物的制作方法
- 用作雄激素受体调节剂的n-(吡啶-4-基)-2-苯基丁酰胺的制作方法
- 可用作雄激素受体调节剂的四氢咔唑衍生物(sarm)的制作方法
- 作为选择性雄激素受体调节剂的取代的n-芳基吡咯烷化合物的制作方法