用作蛋白质聚集抑制剂的杂芳基酰胺的制作方法
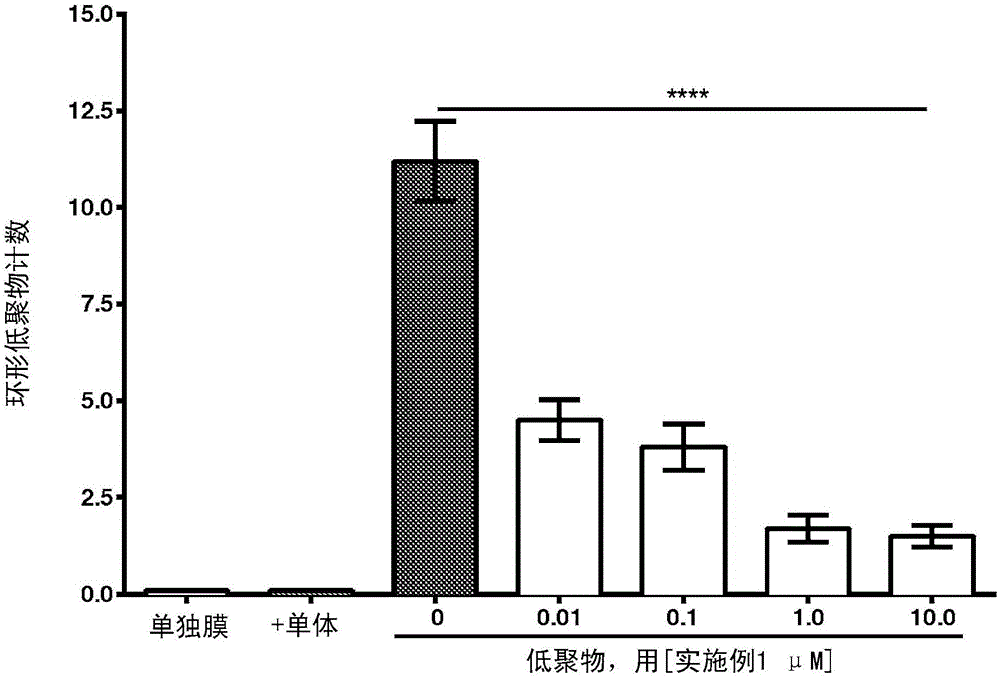
本发明涉及某些杂芳基酰胺衍生物、包含上述物质的药物组合物及其使用方法,所述方法包括防止、反转、延缓或抑制蛋白质聚集的方法和治疗蛋白质聚集相关疾病的方法,所述疾病包括神经变性疾病(如帕金森病、阿尔茨海默病、路易体病、帕金森病痴呆、额颞痴呆、亨廷顿病、肌萎缩侧索硬化、和多系统萎缩症、以及癌症)。序列表申请人在此提交了符合PCT规则13ter.1(a)至(d)和行政指令,部分208和附录C的标准的序列表(通过EFS-WEB电子提交)。申请人声明以ASCII.txt格式的计算机可读形式记录并在此提交的信息并不包括超过提交的国际申请的公开内容的内容。申请人要求考虑进入在此通过EFS-WEB提交的按照Rule5.2(a)作为本申请部分的序列表。txt文件标记为699462000840SeqList。在2015年1月27日产生国际申请的序列表格式部分并且含有645个字节。背景老龄化群体的神经变性疾病(如阿尔茨海默病(AD)、帕金森病(PD)和额颞痴呆(FTD))仅在美国和欧盟就影响了超过2000万人并成为老年人死亡的主要原因之一。这些神经变性疾病之间的共同特点是蛋白质长期积累成神经毒性聚集体。各疾病的特征是受影响的特定神经元群体、涉及的特定蛋白质聚集体和神经元变性所导致的临床特征。研究表明,蛋白质聚集的初始阶段涉及靶蛋白的突变或翻译后修饰(如亚硝化、氧化),其继而生成异常构象,促进与类似错误折叠的蛋白质发生相互作用。异常蛋白随后聚集以形成二聚体、三聚体和更高级的多聚体(也称作“可溶性低聚体”),其可能破坏突触功能。此外,随后聚集体会固定在细胞膜中并形成球形低聚体(其进而会在膜中形成孔)和/或原纤维或纤维。这些较大的不溶性纤维可能用作生物活性低聚体的储器。多种证据支持以下见解:蛋白质聚集体的累进积聚与神经变性疾病的发病有因果关系。多种其他蛋白质可在神经变性疾病的患者脑部积聚,如α-突触核蛋白、Aβ蛋白、Tau和TDP43。这些患者的认知障碍与新皮层和边缘系统突触损失紧密相关并且增加的蛋白质聚积水平可能导致这些突触损失。许多研究聚焦于通过α-突触核蛋白聚积的详细机制并且其他淀粉样前体蛋白(APP)代谢物导致突触损伤和神经变性。许多研究支持了所有聚集体,也称为低聚体的形成在神经毒性中起主要作用的假设。这些肽低聚体可组织成二聚体、三聚体、四聚体、五聚体和其他更高级的阵列,其可形成环形结构。高水平的这类低聚体预示着患者痴呆和突触损失。因为证据表明低聚体而非较小的前体纤维是毒性物质,以特异性方式靶向这些早期聚积过程的化合物可用作PD、AD和相关病症的潜在新疗法。各种神经变性疾病包括基于神经毒性蛋白质聚集体的累积。在特发性帕金森病(IPD)、路易体痴呆(LBD)、帕金森病痴呆(PDD)、和多系统萎缩症(MSA)中,神经毒性聚集体由α-突触核蛋白(SYN)组成,其是正常条件下处于胞内的突触蛋白。在FTD和肌萎缩侧索硬化(ALS)中,神经毒性聚集体源自其他胞内蛋白(如tau、TDP-43或SOD1)。对于特定疾病(如AD),SYN与主要蛋白质聚集(例如,Aβ蛋白)。在亨廷顿氏病中,聚集体从Htt蛋白的切割产物形成。癌症中,尤其是黑素瘤癌细胞中也涉及α-突触核蛋白的累积。Pan等,PLoSOne2012,7(9),e45183。因此,抑制这类累积的化合物可用于治疗各种癌症,包括黑素瘤。这些蛋白质累积过程中显示两种机制。在第一种机制中,错误折叠和/或聚集的蛋白质固定至多种细胞膜结构。错误折叠或聚集的分子与质膜或细胞器(如线粒体或溶酶体)膜的结合会干扰蛋白质转录、自体吞噬、线粒体功能和孔形成。例如,神经毒性SYN通过突触核蛋白C端区域的特定部分聚集并与细胞膜中的脂质相互作用。结合至该区域的化合物可抑制蛋白质-蛋白质或蛋白质-脂质相互作用并因此用于阻碍SYN或其他蛋白质的神经毒性低聚化及其与膜相互作用。在第二过程中,聚集的蛋白质从固定的亚基中释放并传播至相邻细胞。随后,这种毒性蛋白聚集体的细胞至细胞的传播可能是神经变性的解剖学进展和症状恶化的原因。与靶蛋白相互作用的小分子药物可限制释放和/或传播,从而减少聚集蛋白质的神经毒性作用。蛋白质聚积的抑制剂的化合物描述于PCT公开号WO2011/084642、WO2013/148365、WO2013/134371和PCT申请号PCT/US2013/050719。仍然需要具有所需药物特性的蛋白质聚集抑制剂。本发明中,已发现某些杂芳基酰胺化合物具有蛋白质聚集调节活性。技术实现要素:在一个方面,本发明涉及下式(I)的化学实体:其中,R1是H、卤素、C1-4烷基或CF3;R2是H、-CF3、或未取代的或者被卤素或-CF3取代的C1-4烷基;A是5元杂芳基环;Y不存在或者是C1-4亚烷基;R3和R4连同与其相连的氮一起形成未取代的或被C1-4烷基取代的单环杂环烷基环;或者其中Y是C1-4亚烷基,R3和Y连同与R3相连的氮一起形成单环杂环烷基环,并且R4是H或C1-4烷基;或其药学上可接受的盐。在某些实施方式中,式(I)的化合物是选自下文详述中所描述或列举的种类的化合物。在另一个方面,本发明涉及一种药物组合物,所述药物组合物包括至少一种式(I)的化合物或其药学上可接受的盐。本发明所述的药物组合物还可包含药学上可接受的赋形剂。本发明还涉及用作药物的式(I)的化合物或其药学上可接受的盐。在另一个方面,本发明涉及治疗与蛋白或肽聚集相关的神经变性疾病或病症的方法,所述方法包括向需要这类治疗的对象给予有效量的至少一种式(I)的化合物或其药学上可接受的盐。在另一个方面,本发明涉及治疗与蛋白或肽聚集相关的疾病或医学病症的方法,所述方法包括向需要这类治疗的对象给予有效量的至少一种式(I)的化合物或其药学上可接受的盐。本发明还涉及式(I)的化合物在制备用于治疗这类疾病和医学病症的药物中的用途,以及这类化合物和盐用于治疗这类疾病和医学病症的用途。在另一个方面,本发明涉及干扰细胞中蛋白质或肽聚集的累积或防止、延缓、反转或抑制细胞中蛋白质或肽聚集体的方法,所述方法包括将细胞与有效量的至少一种式(I)的化合物或其盐和/或与至少一种本发明的药物组合物接触,其中所述接触是体外、离体或体内接触。从下面的详述和本发明的实践中可以很容易地了解本发明的其他实施方式、特征和优点。为简便起见,该说明书中引用的出版物的内容(包括专利)通过引用纳入本文。附图的简要说明图1显示了生物实施例3的结果。在图1A中,Y轴(I/Io)是在脂膜存在(I)或不存在(Io)的情况下ASYN的(平均残留3-23)异核单量子相干(HSQC)光谱信号强度的比率。在图1B中,ASYN残基3-23的平均I/Io比率以添加的实施例1的浓度的函数作图。图2显示了如生物实施例4所述,在存在和不存在实施例1的情况下,ASYN低聚体的电子显微图像定量。图3显示了实施例1对表达GFP-标记的人ASYN的B103成神经细胞瘤细胞中ASYN的累积,如生物实施例5所述。图4显示了生物实施例6A的结果,以及圆点束任务模型中1mg/kg和5mg/kg剂量的实施例1对转基因小鼠的影响。图5显示了使用A11抗体对大脑和海马区脑部匀浆物的生物实施例6A点印迹分析的结果。图6显示了生物实施例6B的结果和实施例1对种系61ASYN转基因小鼠中皮质神经毡和神经细胞体中ASYN免疫标记的影响。图6A反映了ASYN神经毡臂,并且图6B反映了研究的神经细胞体臂。图7显示了生物实施例6B的结果以及实施例1对包括酪氨酸羟化酶(图7A)、NeuN(图7B)、和胶质细胞原纤维酸性蛋白(GFAP;图7C)的肾病变性相关标志物的免疫标记的影响。图8显示了生物实施例6的结果以及使用圆点束表现测试的实施例1对种系61ASYN转基因小鼠感觉运动损伤的影响。图9显示了生物实施例7A的结果以及实施例1对种系61ASYN转基因小鼠的粪便勃利(fecalboli)计数的影响。图10显示了生物实施例7B的结果以及实施例1对种系61ASYN转基因小鼠中ASYN的心脏水平的影响。图11显示了生物实施例7C的结果以及实施例1对PDNG78转基因小鼠视网膜中含ASYN-GFP的图像面积百分比的影响。图12显示了生物实施例7C的结果以及实施例1对PDNG78转基因小鼠中血管周围和神经末端GFP标记的影响。发明详述在进一步描述本发明之前,应理解,本发明不限于所述的具体实施方式,因为它们当然可能变化。还应理解,本文所用术语的目的仅是描述具体实施方式,不用来构成限制,因为本发明的范围仅受所附权利要求书的限制。除非另有说明,本文所用的所有科技术语与本发明所属领域普通技术人员所理解的通常含义相同。本文中引用的所有专利、申请、公开申请和其他出版物都全文参考结合于本文。如果该部分中的定义与引用纳入本文的专利、申请和其他出版物中所列的定义相反或不一致,该部分中的定义将压倒引用纳入本文的定义。本文和所附权利要求书所用的单数形式“一个”、“一种”和“所述”包括复数指示物,除非上下文另有明确说明。还应注意到,权利要求书可撰写成排除任何任选要素。同样,这种说明应用作引用权利要求元素相关地使用这类排除性术语,如“只有”、“仅仅”等,或使用“负”限制的前提基础。本文所用术语“包含”、“含有”和“包括”以其开放、非限定的含义使用。为提供更简明的描述,本文中的一些定量表达前不使用术语“约”。应理解无论是否明确地使用术语“约”,本文中的每个含量均表示实际给定的数值,且其还表示基于本领域普通技术所能合理推断的给定数值的近似值,包括由于实验和/或测量条件产生的这类给定数值的等同值和近似值。无论何时以百分比表示产率时,这类产率表示在特定的化学计量比条件下用于计算产率的实体质量与同一实体所能获得的最大量的比值。百分比形式的浓度表示质量比率,除非另有说明。除非另有说明,本文所用的所有科技术语与本发明所属领域普通技术人员所理解的通常含义相同。虽然也可采用与本文所述类似或等同的任何方法和材料实施或测试本发明,但以下描述优选的方法和材料。本文提及的所有出版物均通过引用纳入本文,与所引用出版物相关联来公开和描述这些方法和/或材料。除非另有说明,本发明实施方式的方法和技术一般遵循本领域熟知的传统方法进行并如若干一般的或更特定的参考文献中所述,所述参考文献贯穿本说明书引用和讨论。参见,例如,Loudon,OrganicChemistry(《有机化学》),第四版;纽约:牛津大学出版社(OxfordUniversityPress),2002,第360-361、1084-1085页;Smith和March,March′sAdvancedOrganicChemistry:Reactions,Mechanisms,andStructure(《麻尺高级有机化学:反应、机制和结构》),第五版,韦利科学公司(Wiley-Interscience),2001。本文用于命名主题化合物的命名法在本文的实施例中说明。该命名法一般采用市售可得的AutoNom软件(加利福尼亚州圣里安德鲁的MDL公司),版本12.0.2获得。应理解,为清楚起见,在单独的各实施方式的内容中描述的本发明的一些特征还可以合并在单个的实施方式中提供。反之,在单个实施方式的内容中简短描述的本发明的各特征也可以单独提供或以任何合适的子组合的形式提供。与由可变形式代表的化学基团相关的实施方式的所有组合均特定包含在本发明范围内并由本文公开,形同本文单独且明确地公开每个和各个组合以至此类组合包含自身为稳定化合物的化合物(即,可分离、表征和检测生物学活性的化合物)。此外,描述此类可变形式的实施方式中所列的化学基团的所有子组合也具体包含在本发明范围内并由本文公开,形同本文单独且明确地公开化学基团的每个和各个此类子组合。代表性实施方式在式(I)的一些实施方式中,R1是H、氟、氯、溴、甲基、以及、丙基、异丙基、丁基、异丁基、仲丁基、或叔丁基。在其它实施方式中,R1是H或氟。在其它实施方式中,R1是H。在其它实施方式中,R1是氟。在一些实施方式中,R2是H、-CF3,或者是甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、或叔丁基,其各自是未取代的或被氟、氯、溴或-CF3取代。在其它实施方式中,R2是H。在其他实施方式中,R2是-CF3或者是被卤素或-CF3任选取代的C1-4烷基。在其他实施方式中,R2是未取代的或被氟或-CF3取代的C3-4烷基。在其它实施方式中,R2是丁基。在其他实施方式中,R2是被-CF3取代的丙基。在一些实施方式中,R2处于“R”立体化学构型。在其他实施方式中,R2处于“S”立体化学构型。在其他实施方式中,式(I)的化合物是在R2位置处的立体化学混合物。在其他实施方式中,R2基本是“R”或基本是“S”立体化学构型。在一些实施方式中,A是具有2个或3个杂原子环原子的5-元杂芳基环。在其他实施方式中,A是具有2个不相邻杂原子环原子的5-元杂芳基环。在其他实施方式中,A是噻唑、噻二唑、噁唑、咪唑或三唑。在其他实施方式中,A是噻唑或噻二唑。在其他实施方式中,A是噻唑。在一些实施方式中,Y不存在。在其他实施方式中,Y是-CH2-、-CH2CH2-、-CH(CH3)-、-(CH2)3-、-C(CH3)2-、-(CH2)4-、-CH((CH2)2CH3)-、-CH(CH(CH3)2)-、-CH(CH2CH3)CH2-、-CH(CH3)CH(CH3)-、-CH(CH3)(CH2)2-、或-CH2CH(CH3)CH2-。在其他实施方式中,Y是-CH2-、-CH2CH2-或-CH(CH3)-。在其他实施方式中,Y是-CH2CH2-。在一些实施方式中,R3和R4连同与其相连的氮一起形成单环杂环烷基环,其是未取代的或被C1-4烷基取代。在其他实施方式中,R3和R4连同与其相连的氮一起形成氮杂环丁烷、吡咯烷、哌啶、氮杂哌嗪、吗啉、硫代吗啉、或1,1-二氧代-硫代吗啉,各自是未取代的或被C1-4烷基取代。在其它实施方式中,R3和R4连同与其相连的氮一起形成哌嗪、吗啉、或吡咯烷,各自是未取代的或被C1-4烷基取代。在其它实施方式中,R3和R4连同与其相连的氮一起形成哌嗪或吗啉,各自是未取代的或被C1-4烷基取代。在其它实施方式中,R3和R4连同与其相连的氮一起形成哌嗪,其是未取代的或被C1-4烷基取代。在其它实施方式中,R3和R4连同与其相连的氮一起形成哌嗪或4-甲基-哌嗪。在其中Y是C1-4亚烷基的其他实施方式中,R3和Y连同与R3相连的氮一起形成单环杂环烷基环,并且R4是H或C1-4烷基。在其他实施方式中,Y和R3连同与R3相连的氮一起形成吡咯烷或哌嗪。在其它实施方式中,R4是H或甲基。在一些实施方式中,R1是H,R2是H或C1-4烷基(或者是H,或者是C3-4烷基),A是噻唑,Y不存在或者是亚乙基(或者不存在,或者是亚乙基),并且R3和R4连同与它们相连的氮一起形成N-甲基哌嗪。在其他实施方式中,式(I)化合物选自:及其药学上可接受的盐。在其他实施方式中,式(I)化合物选自:及其药学上可接受的盐。在一些实施方式中,式(I)的化合物选自实施例1-11,及其药学上可接受的盐。在其他实施方式中,化合物是盐酸盐形式。在其他实施方式中,化合物是实施例27或28,或其药学上可接受的盐。在其他实施方式中,式(I)的R2处于(S)立体化学构型。在其他实施方式中,式(I)的R2处于(R)立体化学构型。化学定义术语“烷基”表示主链上具有1至12个碳原子的直链或支链烷基基团。烷基基团的示例包括甲基(Me)、乙基(Et)、正丙基、异丙基、丁基、异丁基、仲丁基、叔丁基(tBu)、戊基、异戊基、叔戊基、己基、异己基以及本领域普通技术中的基团,且本文提供的教导被认为相当于任一前述实施例。术语“杂芳基”表示单环、稠合双环或稠合多环的芳族杂环(环结构含有选自碳原子和至多四个杂原子(选自氮、氧和硫)的环原子),每个杂环含有3至12个环原子。杂芳基基团的示范性示例包括以下实体(以适当连接部分的形式):术语“卤素”指氯、氟、溴或碘。术语“卤代”指氯、氟、溴或碘。术语“氧代”代表羰基氧。例如,被氧代取代的环戊基是环戊酮。本领域技术人员应认识到上文所列或所示的种类不是穷举性的,也可选择这些定义的术语范围内的其他种类。术语“取代的”表示特定基团或部分带有一个或多个取代基。术语“未取代的”表示特定基团不带有取代基。术语“任选取代的”表示特定基团是未取代的或被一个或多个取代基取代。在使用术语“取代的”描述结构系统时,取代可发生在系统上任意价态允许的位置上。本文所述的任何式旨在表示该结构式的化合物及某些变体或形式。例如,本文的式指代包括外消旋形式,或者一种或多种对映体、非对映异构体、几何异构体、或其混合物。另外,本文的任何式也指代水合物、溶剂合物、或该化合物的所晶型物,或其混合物。本文给出的任何化学式也旨在代表化合物的未标记的形式以及同位素标记形式。同位素标记的化合物具有本文给定化学式所描述的结构,不同之处在于一个或多个原子被具有选择的原子质量或质量数的原子代替。可掺入本发明化合物的同位素的示例包括氢、碳、氮、氧、磷、氟、氯和碘的同位素,分别例如2H、3H、11C、13C、14C、15N、18O、17O、31P、32P、35S、18F、36Cl和125I。这类同位素标记的化合物可用于代谢研究(优选使用14C)、反应动力学研究(使用例如2H或3H)、包括检测或成像技术(如正电子发射断层成像术(PET)或单光子发射计算体层成像术(SPECT))的药物或底物组织分布试验、或对患者的放射性治疗中。具体而言,18F或11C标记的化合物对于PET或SPECT研究是特别优选的。可如Brooks,D.J.,“ositronEmissionTomographyandSingle-PhotonEmissionComputedTomographyinCentralNervousSystemDrugDevelopment(中枢神经系统药物开发中的正电子发射断层成像术和单光子发射计算体层成像术)”NeuroRx2005,2(2),226-236和本文所引用的参考文献所述进行PET和SPECT研究。另外,用重同位素如氘(即2H)取代能提供因代谢稳定性较高(例如体内半衰期延长或剂量要求降低)而产生的某些治疗优势。本发明的同位素标记化合物及其前药通常可通过进行下面方案或实施例和制备中公开的方法来制备,所述方法是用易于获得的同位素标记试剂取代非同位素标记试剂。在本文中对一类取代基使用时,命名法“Ci-j”(其中i>i)表示独立地实现从i至i(包括i和i)的每一个碳原子数目的本发明实施方式。例如,术语C1-3独立地表示含有一个碳原子(C1)的实施方式、含有两个碳原子(C2)的实施方式和含有三个碳原子(C3)的实施方式。本文所提到的任意双取代基包括多种连接可能性,其中允许多于一种的该可能性。例如,双取代基-A-B-(其中A≠B)在本文中表示A连接至第一取代原子且B连接至第二取代原子的双取代基,也表示A连接至第二取代原子且B连接至第一取代原子的双取代基。本发明还包括式(I)所代表化合物的药学上可接受的盐,优选上文所述的那些以及本文所示的特定化合物,以及包括该盐的药学组合物,以及使用该盐的方法。“药学上可接受的盐”旨在表示本文所示化合物的游离酸或碱的盐,其没有毒性、生物学上可以容忍或者生物学上适于向对象给药。通常参见,S.M.Berge等,“PharmaceuticalSalts(《药用盐》)”J.Pharm.Sci.,1977,66,1-19。优选的药学上可接受的盐是药学上有效并且适于接触对象组织而没有过多的毒性、刺激、或过敏反应的那些。本文所述的化合物可具有足够酸性的基团、足够碱性的基团、两种类型的官能团、或超过一种各类型,并且因此与多种无机或有机碱,以及无机和有机酸反应以形成药学上可接受的盐。药学上可接受的盐的示例包括硫酸盐、焦硫酸盐、硫酸氢盐、亚硫酸盐、亚硫酸氢盐、磷酸盐、磷酸一氢盐、磷酸二氢盐、偏磷酸盐、焦磷酸盐、氯化物、溴化物、碘化物、乙酸盐、丙酸盐、癸酸盐、辛酸盐、丙烯酸盐、甲酸盐、异丁酸盐、己酸盐、庚酸盐、丙炔酸盐、草酸盐、丙二酸盐、琥珀酸盐、辛二酸盐、癸二酸盐、富马酸盐、马来酸盐、1,4-丁炔二酸盐、1,6-己炔二酸盐、苯甲酸盐、氯苯甲酸盐、甲基苯甲酸盐、二硝基苯甲酸盐、羟基苯甲酸盐、甲氧基苯甲酸盐、邻苯二甲酸盐、磺酸盐、甲基磺酸盐、丙基磺酸盐、苯磺酸盐、二甲苯磺酸盐、萘-1-磺酸盐、萘-2-磺酸盐、苯基乙酸盐、苯基丙酸盐、苯基丁酸盐、柠檬酸盐、乳酸盐、γ-羟基丁酸盐、乙醇酸盐、酒石酸盐和扁桃酸盐。其它合适的药学上可接受的盐的列表可见于Remington′sPharmaceuticalSciences(《雷明顿药物科学》),第17版,宾夕法尼亚州伊斯顿的马克出版公司(MackPublishingCompany),1985。对于含有碱性氮的式(I)的化合物,可通过本领域中可采用的任意适当方法制备药学上可接受的盐,例如使用酸处理游离碱,例如使用无机酸(如盐酸、氢溴酸、硫酸、氨基磺酸、硝酸、硼酸、磷酸等)或者使用有机酸(如乙酸、苯乙酸、丙酸、硬脂酸、乳酸、抗坏血酸、马来酸、羟基马来酸、羟乙磺酸、琥珀酸、戊酸、富马酸、丙二酸、丙酮酸、草酸、乙醇酸、水杨酸、油酸、棕榈酸、月桂酸、吡喃糖苷酸(如葡萄糖醛酸或半乳糖醛酸)、α-羟基酸(如扁桃酸、柠檬酸或酒石酸)、氨基酸(如天冬氨酸或谷氨酸)、芳族酸(如苯甲酸、2-乙酰氧基苯甲酸、萘甲酸或肉桂酸)、磺酸(如十二烷基磺酸、对甲苯磺酸、甲磺酸或乙磺酸))或任意相容的酸混合物(如本文实施例中所述)以及任意本领域普通技术中视为等同物或可接受的取代物的其他酸及其混合物。本发明还涉及式(I)的化合物的药学上可接受的前药,以及使用该药学上可接受的前药的治疗方法。术语“前药”表示指定的化合物的前体,在向对象给药后其在体内通过化学或生理过程(如溶剂分解或酶切)或者在生理条件下生成化合物(例如将前药置于生理pH下使其转化为式(I)的化合物)。“药学上可接受的前药”指没有毒性、生物学上可以容忍且生物学上适于向对象给药的前药。选择和制备适当前药衍生物的示范性步骤描述于例如“DesignofProdrugs(《前药设计》)”,H.Bundgaard编,埃尔斯威尔出版社(Elsevier),1985。本发明还涉及式(I)的化合物的药物活性代谢物,以及该代谢物在本发明方法中的用法。“药物活性代谢物”表示式(I)的化合物及其盐在体内的药物活性代谢产物。可使用本领域已知或可采用的常规技术确定化合物的前药和活性代谢物。参见例如Bertolini等,J.Med.Chem.1997,40,2011-2016;Shan等,J.Pharm.Sci.1997,86(7),765-767;Bagshawe,DrugDev.Res.1995,34,220-230;Bodor,Adv.DrugRes.1984,13,255-331;Bundgaard,DesignofProdrugs(《前药设计》)(埃尔斯威尔出版社(ElsevierPress),1985);以及Larsen,DesignandApplicationofProdrugs(《前药设计与应用》)、DrugDesignandDevelopment(《药物设计与开发》)(Krogsgaard-Larsen等编,哈伍德学术出版公司(HarwoodAcademicPublishers),1991)。药物组合物为治疗目的,包括本文所述化合物的药物组合物还可包括一种或多种药学上可接受的赋形剂。药学上可接受的赋形剂指没有毒性且生物学上适于向对象给药的物质。这类赋形剂促进本文所述化合物的给药过程且与活性成分相容。药学上可接受的赋形剂的示例包括稳定剂、润滑剂、表面活性剂、稀释剂、抗氧化剂、粘结剂、着色剂、膨胀剂、乳化剂或调味剂。在优选实施方式中,该发明的药物组合物是无菌组合物。可使用本领域技术人员已知或可以使用的复合技术制备药物组合物。本发明也涉及无菌组合物,包括符合决定该组合物的国家和当地规定的组合物。根据本领域中用于多种剂型制备的常规方法,本文所述药物组合物和化合物可配制为适当药物溶剂或载剂中的溶液剂、乳剂、混悬剂或分散剂,或者丸剂、片剂、锭剂、栓剂、囊剂、糖衣剂、颗粒剂、粉末剂、用于重建的粉末剂或连同固态载剂的胶囊剂。本发明的药物组合物可以通过适当的递送途径给予,如口服、胃肠外、直肠、经鼻、局部或经眼途径,或通过吸入。优选地,该组合物配制为用于静脉内或口服给药。对于口服给药,本发明的化合物可以固体形式(如片剂或胶囊剂)或溶液剂、乳剂或混悬剂的形式提供。为制备口服组合物,可配制本发明的化合物以形成例如每天约0.01至约50mg/kg、或每天约0.05至约20mg/kg、或每天约0.1至约10mg/kg的剂量。其他剂量包括每天约0.1mg至1g,每天约1mg至约10mg,每天约10mg至约50mg,每天约50mg至约250mg、或每天约250mg至1g。口服片剂可包括与相容的药学上可接受的赋形剂(如稀释剂、崩解剂、粘结剂、润滑剂、甜味剂、调味剂、着色剂和防腐剂)混合的活性成分。合适的惰性填料包括碳酸钠和碳酸钙、磷酸钠和磷酸钙、乳糖、淀粉、糖、葡萄糖、甲基纤维素、硬脂酸镁、甘露醇、山梨糖醇等。示例性液体口服赋形剂包括乙醇、甘油、水等。淀粉、聚乙烯吡咯烷酮(PVP)、淀粉乙醇酸钠、微晶纤维素和海藻酸是示例性崩解剂。结合剂可包括淀粉和明胶。如果存在,润滑剂可以是硬脂酸镁、硬脂酸或滑石。如果需要,可以使用某种材料(如单硬脂酸甘油酯或二硬脂酸甘油酯)包被片剂以延缓胃肠道中的吸收,或者使用肠溶衣包被。用于口服给药的胶囊包括硬和软明胶胶囊。为制备硬明胶胶囊,可将活性成分与固体、半固体或液体稀释剂混合。软明胶胶囊可以通过将活性成分与水、油(如花生油或橄榄油)、液体石蜡、短链脂肪酸的单和二甘油酯的混合物、聚乙二醇400或丙二醇混合的方式制备。用于口服给药的液体可以是混悬剂、溶液剂、乳剂或糖浆剂的形式,或者可以是临用前用水或其他合适载剂重建的干燥产品。这类液体组合物可选包含:药学上可接受的赋形剂,如助悬剂(例如山梨糖醇、甲基纤维素、藻酸钠、明胶、羟乙基纤维素、羧甲基纤维素、硬脂酸铝凝胶等);非水性载剂,例如油(例如杏仁油或分级椰子油)、丙二醇、乙醇或水;防腐剂(例如对羟基苯甲酸甲酯或对羟基苯甲酸丙酯或山梨酸);润湿剂(如卵磷脂);以及(如果需要)调味剂或着色剂。本发明的组合物可配制为栓剂用于直肠给药。对于胃肠外使用(包括静脉内、肌肉内、腹膜内、鼻内或皮下途径),本发明的试剂可以无菌水性溶液剂或混悬剂,缓冲至合适pH和等渗性或者以胃肠道外可接受的油的形式提供。合适的水性载剂包括林格氏溶液和等渗氯化钠。这类形式可以是单位剂量形式(如安瓿或一次性注射设备)、多剂量形式(如可以从中取出合适剂量的药瓶)或固体形式或可用于制备可注射制剂的预浓缩液。在数分钟至数天的时间内,示范性输注剂量的范围是约1至1000μg/kg/分钟的与药物运载体混合的试剂。对于经鼻、吸入或口服给药,可使用例如也包含合适运载体的喷雾制剂给予本发明的药物组合物。对于局部使用,优选将本发明的化合物配制为乳膏或软膏或适用于局部给药的类似载剂。对于局部给药,可将本发明的化合物与药物运载体混合,浓度为药物占载剂的约0.1%至约10%。另一个给予本发明的试剂的模式是利用贴片制剂以达到透皮递送的效果。本文所用术语“治疗”或“处理”包括“预防性”和“治疗性”治疗。“预防性”治疗指推迟疾病、疾病症状或医学病症的发展、抑制可能出现的症状或降低疾病或症状发展或复发的风险。“治疗性”治疗包括降低已有疾病、症状或病症的严重程度或抑制其恶化。因此,治疗包括改善或防止已有疾病症状的恶化、防止出现其他症状、改善或防止症状的根本性系统原因、抑制失调或疾病,例如阻止失调或疾病的发展、减轻失调或疾病、促使失调或疾病退行、减轻疾病或失调引起的病症或停止疾病或失调的症状。术语“对象”指需要该治疗的哺乳动物患者,例如人。示例性的特征为蛋白质聚积的神经变性疾病包括阿尔茨海默病、帕金森病、额颞痴呆、路易体痴呆(路易体病)、帕金森病痴呆、多系统萎缩症、肌萎缩侧索硬化、和亨廷顿病,以及癌症和炎性疾病。在一个方面,本发明的化合物和药物组合物特异性靶向α-突触核蛋白、β-淀粉样蛋白和/或tau蛋白聚集体。因此,这些化合物和药物组合物可用于防止、反转、延缓或抑制α-突触核蛋白、β-淀粉样蛋白和/或tau蛋白的聚集,并用于本发明的方法中以治疗聚集(如α-突触核蛋白、β-淀粉样蛋白和/或tau蛋白的聚集)相关或引起的神经变性疾病。优选地,本发明的方法靶向与α-突触核蛋白、β-淀粉样蛋白和/或tau蛋白的聚集相关的神经变性疾病。在优选的实施方式中,治疗方法靶向帕金森病、阿尔茨海默病、路易体病、或多系统萎缩症。在其他实施方式中,方法靶向癌症或黑色素瘤。本发明的化合物、组合物和方法还可用于减轻蛋白质聚集继发的有害作用,如神经元细胞死亡。在一些方面中,本发明的化合物、组合物和方法用于靶向α-突触核蛋白(SYN)聚集。在另一个的方面,本发明的化合物、组合物和方法用于靶向Aβ聚集。在本发明的抑制性方法中,“有效量”指足以减少、延缓或反转蛋白质或肽聚集的量。可通过如下文所述的常规分析方法对聚集的量进行测量。这类修饰可用于多种环境,包括体外试验。在该方法中,所述细胞优选是神经细胞。在根据本发明的治疗方法中,“有效量”指足以使需要这类治疗的对象获得所需治疗益处的量或剂量。本发明的化合物的有效量或剂量可通过常规方法(如建模、剂量递增或临床试验)确定,其中考虑常规因素,例如给药或药物递送的模式或途径、试剂的药代动力学、感染的严重程度和过程、对象的健康状态、病情和体重以及主治医生的判断。示例性剂量的范围为每天每千克对象体重约1ug至2mg活性试剂,优选约0.05至100mg/kg/天、或约1至35mg/kg/天、或约0.1至10mg/kg/天。在其他实施方式中,示例性剂量的范围是约1mg至约1g/天,或约1-500、1-250、1-100、1-50、50-500、或250-500mg/天。总剂量可以单个或分开的剂量单元(例如BID、TID、QID)给予。一旦患者的疾病出现改善,即可调节剂量用于预防性或维持治疗。例如,给药的剂量或频率或两者可以随着症状变化降至保持所需治疗或预防效果的水平。当然,如果症状已减轻至合适水平,可以停止治疗。但任何症状复发时,患者可以要求长期基础上的间歇治疗。患者可能还需要长时程基础上的长期治疗。药物组合本发明的化合物可与一种或多种神经退行性疾病的治疗中的其他活性成分联用,用于药物组合物或方法。用于癌症应用的其他活性成分包括减轻癌症化疗剂的不良影响的其他癌症治疗药或试剂。这类组合可用于增加效力、改善其他疾病症状、减少一种或多种副作用或减少所需的本发明化合物剂量。其他活性成分可以独立于本发明的化合物的药物组合物形式给予,或者可与本发明的化合物一起包含在单个药物组合物中。其他活性成分可以与本发明的化合物同时给予、在其之前给予或在其之后给予。包含其他活性成分的组合试剂是那些已知或被发现能够有效治疗神经变性疾病的成分,包括对疾病相关的另一个靶标有活性的那些,诸如但不限于:a)解决蛋白质错误折叠的化合物(如减少这些蛋白质生成、提高其清除或改变其聚集和/或传播的药物);b)治疗该疾病症状的化合物(例如多巴胺替代疗法);以及c)通过互补机制作为神经保护剂的药物(例如靶向自体吞噬的那些药物,抗氧化的那些药物以及通过其他机制作用的那些药物(如腺嘌呤A2A拮抗剂))。例如,本发明的组合物和制剂以及治疗方法还可包括其他药物或药品,例如用于治疗或缓解蛋白质聚集(例如突触核蛋白、β-淀粉样蛋白和/或tau蛋白的聚集)相关或引起的神经变性疾病(例如帕金森病、阿尔茨海默病(AD)、路易体病(LBD)和多系统萎缩症(MSA)或相关症状或病症)的其他活性剂。例如,本发明的药物组合物还可包括一种或多种该活性剂,且治疗方法还可包括给予有效量的一种或多种该活性剂。在某些实施方式中,其他活性剂可以是抗生素(例如抗菌或抑菌肽或蛋白质,例如有效对抗革兰氏阳性或阴性细菌的那些)、流体、细胞因子、免疫调节剂、抗炎剂、补体活化剂(如包括胶原样结构域或血纤蛋白原样结构域(如纤维凝胶蛋白(ficolin))、碳水化合物结合结构域等的肽或蛋白质)及其组合。其他活性剂包括用于该组合物和方法的那些,包括多巴胺疗法药物、邻苯二酚-O-甲基转移酶(COMT)抑制剂、单胺氧化酶抑制剂、认知增强剂(如乙酰胆碱酯酶抑制剂或美金刚胺)、腺嘌呤2A受体拮抗剂、β-分泌酶抑制剂或γ-分泌酶抑制剂。在特定实施方式中,本发明的至少一种化合物可在药物组合物或治疗方法中与一种或多种药物联用,所述药物选自:他克林(Cognex)、多奈哌齐(Aricept)、利凡斯的明(Exelon)、加兰他敏(Reminyl)、毒扁豆碱、新斯的明、艾考哌齐(Icopezil)(CP-118954,5,7-二氢-3-[2-[1-(苯基甲基)-4-哌啶基]乙基]-6H-吡咯并-[4,5-f-]-1,2-苯并异唑-6-酮马来酸盐)、ER-127528(4-[(5,6-二甲氧基-2-氟-1-茚酮)-2-基]甲基-1-(3-氟苄基)哌啶盐酸盐)、扎那普齐(zanapezil)(TAK-147;3-[1-(苯基甲基)哌啶-4-基]-1-(2,3,4,5-四氢-1H-1-苯并氮杂-8-基)-1-丙烷富马酸盐)、美曲磷酯(Metrifonate)(T-588;(-)-R-.α.-[[2-(二甲基氨基)乙氧基]甲基]苯并[b]噻吩-5-甲醇盐酸盐)、FK-960(N-(4-乙酰-1-哌嗪基)-对氟苯甲酰胺水合物)、TCH-346(N-甲基-N-2-丙酰联苯并[b,f]氧杂-10-甲胺)、SDZ-220-581((S)-.α.-氨基-5-(膦酰基甲基)-[1,1′-联苯基]-3-丙酸)、美金刚胺(Namenda/Exiba)和1,3,3,5,5-五甲基环己烷-1-胺(Neramexane)、氟比洛芬(tarenfiurbil)(Flurizan)、3-氨基丙烷磺酸(tramiprosate)(Alzhemed)、氯碘喹啉(clioquinol)、PBT-2(8-羟基喹诺酮衍生物)、1-(2-(2-萘基)乙基)-4-(3-三氟甲基苯基)-1,2,3,6-四氢吡啶、石杉碱A、泊替瑞林(posatirelin)、亮丙瑞林(leuprolide)或其衍生物、异丙克兰(ispronicline)、(3-氨基丙基)(正丁基)次膦酸(SGS-742)、N-甲基-5-(3-(5-异丙氧基吡啶基))-4-戊-2-胺(ispronicline)、1-癸基铵(1-decanaminium)、N-(2-羟基-3-磺丙基)-N-甲基-N-辛基-、内盐(zt-1)、水杨酸酯、阿司匹林、阿莫比林、贝诺酯、胆碱水杨酸镁、二氟尼柳、法西阿明、水杨酸甲酯、水杨酸镁、双水杨酯、双氯芬酸、醋氯芬酸、阿西美辛、溴芬酸、依托度酸、吲哚美辛、萘丁美酮、舒林酸、托美汀、布洛芬、卡洛芬、芬布芬、非诺洛芬、氟比洛芬、酮洛芬、酮咯酸、洛索洛芬、萘普生、噻洛芬酸、舒洛芬、甲芬那酸、甲氯灭酸、苯基丁氮酮、阿扎丙宗、安乃近、羟布宗、磺吡酮、吡罗昔康、氯诺昔康、美洛昔康、替诺昔康、塞来昔布、依他昔布、罗美考昔布、帕瑞昔布、罗非昔布、伐地昔布、尼美舒利、芳基烷酸、2-芳基丙酸(profens)、N-芳基邻氨基苯甲酸(灭酸)、吡唑烷衍生物、昔康类、COX-2抑制剂、磺酰苯胺(sulphonanilides)、必需脂肪酸和米诺扎(Minozac)(2-(4-(4-甲基-6-苯基哒嗪-3-基)哌嗪-1-基)嘧啶二盐酸水合物)或其组合物。用于癌症疗法的潜在组合试剂可包括,例如,蛋白质和脂质激酶抑制剂(例如,PI3K、B-raf、BCR/ABL)、放疗增强剂、微管结合剂(例如,紫杉醇、长春碱)、细胞代谢抑制剂、DNA嵌入剂、拓扑异构酶抑制剂(例如,多柔比星)、和DNA烷基化试剂。测定本文所述的化合物可用于研究应用,包括体外、体内或离体实验系统。实验性系统可包括,但不限于,细胞样品、组织样品、细胞组分或细胞组分混合物、完整或部分器官、或生物体。研究应用包括,但不限于,用作测定试剂、生物化学通路解析、或其他试剂对实验系统的解析,在一种或多种本文所述的化合物存在或缺失下。本文所述的化合物也可用于生物化学试验。在一些实施方式中,本文所述的化合物可与来自对象的组织或细胞孵育以评价对象对化合物给予的潜在响应,或确定本文所述的哪种化合物在特定对象或对象组中阐述有关最优效果。一种这类试验包括(a)从对象获取细胞样品或组织样品,其中可测试一种或多种生物标志物的调节,(b)向细胞样品或组织样品给予一种或多种本文所述的化合物;和(c)与给予化合物前的生物标志物的状态相比,确定给予化合物后一种或多种生物标志物的调节量。任选地,在步骤(c)后,试验将包括基于步骤(c)中确定的调节量选择用于治疗与蛋白质聚集相关的疾病或医学病症的化合物的额外步骤(d)。化学合成现在,通过参考用于本文中通用制备方法的示意性合成方案和之后的具体实施例来描述本发明的方法中有用的示例性化学实体。本领域技术人员应认识到,为获得本文中的多种化合物,可对起始材料进行适当选择,从而通过适当地带有或不带有保护的反应方案使用最终所需的取代基以生成所需的产物。或者,可能需要或想要在最终所需的取代基位置上采用可通过反应方案携带并在适当时可被所需取代基替代的合适基团。此外,本领域技术人员应认识到,以下方案中显示的转化可以任意与特定侧基功能相容的顺序进行。通用方案中描述的各反应均优选在约0℃至所用有机溶剂的回流温度下进行。除非另有说明,变量如上文所定义,可参考式(I)。本文所述同位素标记的化合物根据下文所述方法制备,使用适当标记的起始材料。这类材料通常可从放射标记的化学试剂的市场供应商处购得。方案A如方案A所示制备式(I)的化合物。氨基-乙基吲哚衍生物A1市售可得或按照方案B制备。在标准酰胺形成条件下,化合物A1与活化的酰基化合物A2偶联,其中X是例如-OH或-C1,以产生式(I)的化合物。方案B如方案B所示,通过酰基化,之后还原性胺化从甲基-吲哚B1制备取代的吲哚A1。方案C按照方案C制备杂环化合物C4。某些化合物A、C1、A-CO2R(其中R是H或C1-4烷基)、和C2市售可得。在一些实施方式中,杂环A经卤化以形成卤代化合物C1,并且然后酰基化以形成双官能化的化合物C2。在其他实施方式中,化合物A-CO2R经卤化以形成化合物C2。在标准酰胺偶联条件下与胺HNR3R4偶联形成化合物C3。酯C3的水解产生氨基酸C4,其可用于方案A中所示的偶联反应。方案D如方案D所示,用例如多聚甲醛同系化甲基-杂环化合物D1以产生羟乙基化合物D2。羟基活化成例如卤素或甲苯磺酸基,并被HNR3R4替换,产生氨基化合物D3。杂环的酰基化产生酯D4,并且水解生成氨基酸D5。实施例以下实施例用于说明但非限制本发明。本领域技术人员将认识到可通过选择合适的起始材料和试剂来修饰以下合成反应和方案以获得式(I)的其他化合物。实施例1:N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺.步骤1.在0℃下向干1,2-二氯乙烷(80mL)中化合物1A(6g,45.8mmol)的溶液中加入AlCl3(18.3g,137.4mmol)。混合物升温至25℃并且搅拌30分钟。混合物冷却至0℃并且滴加化合物1B(6.2g,51.3mmol)。将混合物在25℃下搅拌48小时。将反应混合物缓慢倒入冰水中并用二氯甲烷(DCM,三次(3x))萃取。用盐水洗涤有机层(3x),在Na2SO4上干燥并浓缩。通过柱(20∶1石油醚/EtOAc)纯化残留物以得到黄色固体的化合物1C(4.8g,49%)。1HNMR(400MHz,CDCl3)δ8.16(brs,1H),7.39(d,J=8.0Hz,1H),7.37(d,J=8.0Hz,1H),7.23(t,J=7.2Hz,1H),7.15(t,J=7.2Hz,1H),7.15(s,1H),3.83(s,2H),2.50(t,J=8.8Hz,2H),1.54-1.63(m,2H),1.24-1.29(m,2H),0.86(t,J=7.6Hz,3H).步骤2.向MeOH(150mL)中化合物1C(4.8g,22.3mmol)的混合物中加入AcONH4(3.35g,44.6mmol)和NaBH3CN(2.8g,44.6mmol)。混合物回流24小时。混合物浓缩,并且残留物溶于DCM(150mL)中,用盐水洗涤(3x),在Na2SO4上干燥,并浓多以得到黄色固体的粗化合物1D(2.4g,50%)。1HNMR(400MHz,CDCl3)δ8.09(s,1H),7.54(d,J=8.0Hz,1H),7.29(d,J=8.0Hz,1H),7.03-7.18(m,2H),6.98(s,1H),3.01-3.03(m,1H),2.89(dd,J=14.0,4.0Hz,1H),2.52(dd,J=14.0,8.8Hz,1H),1.18-1.50(m,8H),0.85(t,J=7.2Hz,3H).步骤3.将内含化合物1E(2.2g,10.0mmol)、N-甲基哌嗪(1.1g,11.0mmol)和K2CO3(3.4g,24.9mmol)的乙腈(70mL)混合物在80℃下搅拌24小时。混合物经浓缩并用H2O稀释并用EtOAc萃取(3x)。对合并的有机层进行干燥并浓缩以获得淡棕色固体状的化合物1G(2.5g,>100%)。1HNMR(400MHz,CDCl3)δ7.85(s,1H),4.28(q,J=7.2Hz,2H),3.58(t,J=5.6Hz,4H),2.50(t,J=5.6Hz,4H),2.33(s,3H),1.32(t,J=7.2Hz,3H).步骤4.向内含化合物1G(2.0g,8.3mmol)的THF(20mL)混合物添加内含NaOH(1.33g,33.2mmol)的H2O(40mL)溶液。将混合物在80℃下搅拌24小时。混合物经浓缩以去除THF并用n-BuOH萃取。有机层经干燥并浓缩以获得白色固体的化合物1H(1.38g,66.7%)。1HNMR(400MHz,MeOD)δ7.55(s,1H),3.49-3.52(m,4H),2.53-2.56(m,4H),2.36(s,3H).步骤5.向内含化合物1H(1.1g,5.1mmol)的CH2Cl2(50mL)和THF(5mL)的混合物中加入化合物1D(2g,8.09mmol),之后加入苯并三唑-1-基-氧三吡咯烷基六氟磷酸鏻(PyBOP,3.18g,6.12mmol)和DIPEA(2.6g,20.4mmol)。在25℃下N2下24小时后,用H2O(40mL)稀释混合物并用DCM萃取(3x)。合并的有机层在Na2SO4上干燥,浓缩并且通过制备型-HPLC(ShimadzuLC-8A制备型HPLC,Luna(2)C18柱,在20分钟内以80mL/分钟NH4OAc中26%-56%乙腈)纯化残留物以得到白色固体的实施例1(576mg,27%)。1HNMR(400MHz,CDCl3)δ8.23(s,1H),7.39(1H,s),7.38(d,J=7.2Hz,1H),7.22(d,J=7.2Hz,1H),7.07-7.21(m,2H),7.06(s,1H),5.50(d,J=8.4Hz,1H),4.40-4.46(m,1H),3.56(t,J=5.2Hz,4H),3.0-3.1(m,2H),2.52(t,J=4.8Hz,4H),2.36(s,3H),1.26-1.60(m,6H),0.88(t,J=7.2Hz,3H)LCMS:100%(M+1+):426.2.替代合成向内含化合物3A(400g,0.98mol,如下所示)和K2CO3(340g,2.5mol)的CH3CN(3L)的混合物中加入化合物1F(197g,1.97mol)。将混合物在80℃下在N2气氛下搅拌12小时。用水(4L)稀释该混合物并用DCM萃取(4Lx3)。用Na2SO4干燥合并的有机层,然后浓缩。用1∶1石油醚/乙酸乙酯洗涤残留物以得到白色固体的实施例1(200g,44%)。向含实施例1(200g,0.47mol)的DCM(2L)溶液中加入含4MHCl的乙酸乙酯(235mL)。混合物在室温下搅拌1小时,浓缩溶剂,并且从甲基叔丁基醚(1L)中重结晶残留物。通过过滤收集残留固体并且在50℃下减压干燥以得到实施例1的HCl盐(210g,92%)。实施例2:N-(2-(1H-吲哚-3-基)乙基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺.步骤1.在0℃下,向含化合物2A(20g,84.7mmol)的四氢呋喃(THF,100mL)溶液中滴加含NaOH(4.1g,102mmol)的H2O(100mL)溶液。将混合物在10℃下搅拌1小时。混合物用HCl(6M)中和并用EtOAc萃取(3x)。对有机层使用盐水洗涤、使用Na2SO4干燥并浓缩以获得黄色固体的化合物2B(17.7g,100%)。1HNMR(400MHz,DMSO-d6)δ8.21(s,1H).步骤2.向含化合物2B(2.6g,12.5mmol)的THF(16mL)的混合物中加入1,1-羰基-二咪唑(2.06g,12.75mmol)。混合物在室温下搅拌1小时,然后用三乙胺(TEA,2.53g,25mmol)和化合物2C(2g,12.5mmol)处理。所得混合物在室温下搅拌12小时。混合物用EtOAc稀释,用1MHCl(2x)和盐水洗涤,然后在Na2SO4上干燥,并浓缩以得到黄色固体的化合物2D(4.41g,>100%)。步骤3.向内含化合物2D(1g,5.7mmol)和K2CO3(1.0g,7.2mmol)的CH3CN(6mL)的混合物中加入N-甲基哌嗪(0.6g,5.7mmol)。将混合物在80℃下在N2气氛下搅拌12小时。加入水并且所述混合物用DCM(5x)萃取。合并的有机层在Na2SO4上干燥并浓缩以得到黄色固体(0.8g),其用甲基叔丁基醚(5mL)洗涤以得到白色固体的实施例2(0.3g,29%)。1HNMR(400MHz,DMSO-d6)δ8.37(m,1H),7.78(s,1H),7.56(d,J=7.6Hz,1H),7.40(d,J=8.4Hz,1H),7.38-7.35(m,2H),7.16(s,1H),7.07(t,J=7.4Hz,1H),6.98(t,J=7.4Hz,1H),3.48-3.44(m,6H),2.90(t,J=7.2Hz,2H),2.42-2.39(m,2H),2.22(s,3H).MS:(M+H+):370.实施例3:N-(1-(1H-吲哚-3-基)己-2-基)-2-(哌嗪-1-基)噻唑-5-羧酰胺.步骤1.向含化合物2B(5.19g,24.96mmol)的THF(40mL)的混合物中加入1,1-羰基-二咪唑(4.04g,24.96mmol)。混合物在室温下搅拌1小时,然后用TEA(7.56g,74.88mmol)和化合物1D(5.4g,24.96mmol)处理。在室温下12小时后,混合物用EtOAc稀释,并用1MHCl(2x)和盐水连续洗涤,在Na2SO4上干燥,并浓缩以得到黄色固体的化合物3A(7.74g,76%)。1HNMR(400MHz,MeOD-d4)δ8.36(d,1H,J=8.4Hz),7.97(s,1H),7.56(d,J=8Hz,1H),7.30(d,J=8Hz,1H),7.06(t,J=7.2Hz,1H),7.05(s,1H),6.95(t,J=7.2Hz,1H),4.31-4.28(m,1H),3.0-2.97(m,1H),1.75-1.50(m,2H),1.45-1.25(m,4H),2.22(t,J=6.8Hz,3H).MS:(M+H+):406,408.步骤2.向内含化合物3A(2g,5.7mmol)和K2CO3(1.77g,12.8mmol)的CH3CN(12mL)的混合物中加入化合物3B(0.92g,4.9mmol)。在80℃下N2下12小时后,加入水(30mL)并用EtOAc萃取混合物(3x)。合并的有机层在Na2SO4上干燥、浓缩、并且通过柱层析(10至67%EtOAc/石油醚)纯化以得到黄色固体的化合物3C(2g,79%)。该化合物无需表征即可直接用于下一步骤。步骤3.用HCl(10mL,4M,甲醇中)处理含化合物3C(1g,2.0mmol)的甲醇(10mL)溶液。在室温下搅拌3小时后,去除溶剂并且将残留物倒入冰水中(20mL),用NaHCO3将pH调整到9,并用EtOAc萃取(2x)。合并的有机层在Na2SO4上干燥并浓缩以得到黄色固体(0.45g)。通过柱色谱(石油醚/DCM/MeOH50/50/0至0/90/10)纯化得到白色固体的实施例3(0.32g,39%)。1HNMR(400MHz,CDCl3)δ8.16(s,1H),7.64(d,J=7.6Hz,1H),7.39-7.36(m,2H),7.18(t,J=7.6Hz,1H),7.13(t,J=7.6Hz,1H),7.05(s,1H),5.53(d,J=8.8Hz,1H),4.46-4.38(m,1H),3.58-3.49(m,4H),3.06-2.96(m,6H),2.05(s,1H),1.65-1.60(m,1H),1.48-1.32(m,5H),0.90-0.86(m,3H).MS:(M+H+):412.实施例4:N-(2-(1H-吲哚-3-基)乙基)-2-(2-(4-甲基哌嗪-1-基)乙基)噻唑-5-羧酰胺.步骤1.将在密封管中的化合物4A(50g,0.5mol)和多聚甲醛(50g)的混合物在140℃下搅拌3小时。通过柱色谱(50%DCM/EtOAc)纯化反应混合物以得到淡黄色油状的化合物4B(27g,41%)。1HNMR(400MHz,CDCl3)δ7.62(s,1H),7.16(s,1H),3.91-3.98(m,2H),3.23-3.28(m,2H).步骤2.在0℃下向内含化合物4B(27g,0.2mmol)的THF(100mL)溶液中添加内含NaOH(16.7g,0.4mol)的水(100mL)溶液。在搅拌10分钟后,分步添加4-甲基苯-1-磺酰氯(59.5g,0.3mol)。混合物升温至室温并总共搅拌2小时。用EtOAc(3x)提取混合物。有机相用盐水洗涤,在Na2SO4上干燥,浓缩,并通过柱色谱(己烷/EtOAc=2∶1)纯化以得到无色油状的化合物4C(32g,54%)。1HNMR(400MHz,CDCl3)δ7.75(2H,d,J=8Hz,2H),7.65(1H,d,J=4Hz,1H),7.32(d,J=8Hz,2H),7.22(d,J=4Hz,1H),4.40(t,J=8Hz,2H),3.80(t,J=8Hz,2H),2.45(s,3H).步骤3.向含化合物4C(32g,0.11mol)的乙腈(300mL)溶液中加入N-甲基哌嗪(16.9g,0.16mol)和Cs2CO3(55g,0.16mol)。混合物在60℃下搅拌过夜,然后过滤混合物并浓缩滤液,通过柱色谱(DCM/MeOH=10∶1)纯化残留物以得到淡黄色油状的化合物4D(14g,47%)。1HNMR(400MHz,CDCl3)δ7.67(d,J=3.2Hz,1H),7.19(d,J=2.4Hz,1H),3.21(t,J=8.0Hz,2H),2.79(t,J=8.0Hz,2H),2.59(s,4H),2.50(s,4H),2.31(s,3H).步骤4.在-78℃下向含化合物4D(14g,66mmol)的无水THF(70mL)溶液中滴加正丁基锂(32mL,80mmol)。混合物在该温度下搅拌30分钟,然后在相同温度下向溶液中滴加氯甲酸甲酯(7.52g,80mmol)。混合物搅拌3小时,在期间升温至室温。混合物用饱和水性NH4Cl(50mL)稀释,用EtOAc萃取,用盐水洗涤,在Na2SO4上干燥,并浓缩以得到化合物4E(14g,78%)。该粗产物无需进一步纯化即可直接用于下一步骤。1HNMR(400MHz,CDCl3)δ8.26(s,1H),3.89(s,3H),3.19(t,J=8.0Hz,2H),2.77(t,J=8.0Hz,2H),2.58(s,4H),2.49(s,4H),2.17(s,3H).步骤5.向内含化合物4E(14g,52mmol)的MeOH(100mL)溶液中添加内含NaOH(3.12mL,78mmol)的水(39mL)溶液。在室温下3小时后,混合物浓缩并且用EtOAc(30mL)洗涤。用1NHCl将水相调整到pH=5-6并且用EtOAc(3x)萃取。合并的有机层用盐水洗涤,在Na2SO4上干燥,并浓缩以得到橙色固体的化合物4F(12g,92%)。该粗产物无需进一步纯化即可直接用于下一步骤。步骤6.内含化合物4F(0.8g,3.1mmol)的DCM(2mL)和THF(10mL)溶液用碘化2-氯-1-甲基-吡啶鎓(Mukaiyama试剂,1.0g,3.8mmol)和N,N-二异丙基乙胺(DIPEA,1.5g,16mmol)处理,然后搅拌10分钟。然后用2-(1H-吲哚-3-基)乙胺(0.5g,3.1mmol)处理混合物并且混合物在50℃下搅拌3小时。滤去所得沉淀并真空浓缩滤液。残留物溶于乙酸乙酯中,用水(10mL)和盐水(10mL)洗涤,用Na2SO4干燥,并浓缩。通过制备型HPLC(ShimadzuLC-8A,GeminiC-18,0.04%水性NH4OH中12-42%CH3CN,在20分钟内,以80mL/分钟)纯化残留物以得到淡黄色固体的实施例4(200mg,16%)。1HNMR(400MHz,CDCl3)δ8.10(s,1H),7.81(1H,s,1H),7.65(d,J=8Hz,1H),7.40(d,J=8Hz,1H),7.21(t,J=8Hz,1H),7.08(s,1H),5.99(brs,1H),3.77(q,J=6.2Hz,2H),3.16(t,J=7.1Hz,3H),3.10(t,J=6.6Hz,2H),2.75(t,J=6.8Hz,2H),2.58(brs,4H),2.49(brs,4H),2.32(s,3H).实施例5:N-(1-(1H-吲哚-3-基)己-2-基)-2-(2-(4-甲基哌嗪-1-基)乙基)噻唑-5-羧酰胺.内含化合物4F(1.2g,5mmol)的DCM(2mL)和THF(10mL)溶液用Mukaiyama试剂(1.65g,6.5mmol)和DIPEA(1.9g,20mmol)处理,然后搅拌10分钟。混合物然后用化合物1D(1.2g,5mmol)处理并在50℃下搅拌3小时。滤去所得沉淀并真空浓缩滤液。残留物溶于乙酸乙酯(30mL)中,用水(10mL)和盐水(10mL)洗涤,用Na2SO4干燥,并浓缩。通过制备型HPLC(ShimadzuLC-8A,GeminiC-18,0.04%水性NH4OH中25-55%CH3CN,在20分钟内,以80mL/分钟)纯化以得到白色固体的实施例5(250mg,14%)。1HNMR(400MHz,CDCl3)δ8.10(s,1H),7.75(s,1H),7.64(d,J=8Hz,1H),7.38(d,J=8Hz,1H),7.19-7.21(m,1H),7.13-7.14(m,1H),5.13(brs,1H),4.41-4.45(m,1H),3.13-3.18(m,1H),3.05-3.09(m,1H),2.74-2.77(m,1H),2.58(brs,4H),2.49(brs,4H),2.32(s,3H),1.63-1.70(m,1H),1.49-1.54(m,1H),1.34-1.39(m,4H),0.89(t,J=4Hz,3H).实施例6:N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噁唑-5-羧酰胺.步骤1.在-60℃下向含化合物6A(15.0g,106mmol)的THF(150mL)溶液中滴加双三甲基硅基氨基锂(lithiumhexamethyldisilazide)(178mL,170mmol)。将溶液在-50℃下搅拌1小时。然后向溶液中加入六氯乙烷(37.8g,160mmol)。将溶液在室温下搅拌12小时。通过加入饱和水性NH4Cl溶液(50mL)终止该反应并用EtOAc(50mL)萃取。有机层分离,在Na2SO4上干燥,真空浓缩,并且用柱层析纯化残留物以得到无色油状的化合物6B(10g,56%)。1HNMR(400MHz,CDCl3)δ7.71(s,1H),3.39(q,J=7.2Hz,2H),1.38(t,J=7.2Hz,3H).步骤2.将内含化合物6B(5.5g,31.3mmol)、N-甲基哌嗪(9.4g,94mmol)和K2CO3(17.3g,125.2mmol)的乙腈(80mL)混合物在80℃下回流加热2小时。混合物用H2O稀释并用EtOAc萃取。分离有机层,使用Na2SO4干燥,并真空浓缩以获得棕色油状的化合物6C(7g,94%)。1HNMR(400MHz,CDCl3)δ7.52(s,1H),4.32(q,J=7.2Hz,2H),3.66(t,J=4.8Hz,4H),2.48(q,J=4.8Hz,4H),2.34(s,3H),1.34(t,J=7.2Hz,3H).步骤3.内含化合物6C(2.0g,8.4mmol)和NaOH(0.33g,8.4mmol)的THF(10mL)和H2O(10mL)混合物在室温下搅拌2小时。真空浓缩混合物以得到黄色固体的化合物6D(3.0g,粗)。1HNMR(400MHz,D2O)δ7.26(s,1H),3.51(s,4H),2.49(s,4H),2.23(s,3H).步骤4.内含化合物6D(2.0g,9.5mmol)、化合物1D(1.64g,7.6mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(EDC,3.63g,19mmol)、1-羟基-苯并三唑(HOBt,2.57g,19mmol)、和TEA(1.92g,19mmol)的DMF(30mL)的混合物在室温下搅拌12小时。用水稀释该混合物并用EtOAc萃取。分离有机层,用水(3x)、盐水洗涤,在Na2SO4上干燥并真空浓缩。用柱色谱(2至10%MeOH/DCM)纯化残留物以得到白色固体的实施例6(220mg,6%)。1HNMR(400MHzCDCl3)δ8.16(s,1H),7.63(d,J=8.0Hz,1H),7.40(s,1H),7.39(t,J=8.0Hz,1H),7.18(d,J=8.0Hz,1H),7.10(t,J=8.0Hz,1H),7.08(s,1H),5.69(d,J=8.8Hz,1H),4.47(t,J=8.4Hz,1H),3.47(d,J=2.0Hz,4H),3.08-2.98(m,2H),2.49(t,J=4.4Hz,4H),2.36(s,3H),1.66-1.51(m,1H),1.51-1.49(m,1H),1.39-1.27(m,4H),0.90(d,J=7.2Hz,3H).实施例7:N-(1-(1H-吲哚-3-基)己-2-基)-5-(4-甲基哌嗪-1-基)-1,3,4-噻二唑-2-羧酰胺.步骤1.内含化合物7A(60g,0.313mol)、K2CO3(130g,0.94mol)、和甲基哌嗪的DMF(300mL)溶液在40℃下搅拌3小时。将反应混合物倒入水中,用CH2Cl2萃取。对有机层使用水洗涤、使用Na2SO4干燥并浓缩以获得黄色固体的化合物7B(58.5g,73%)。1HNMR(400MHz,CDCl3)δ4.37-4.47(m,2H),3.60-3.71(m,4H),2.47-2.60(m,4H),2.34(s,3H),1.35-1.47(m,3H).步骤2.在室温下向内含化合物7B(5.0g,19.53mmol)的THF(30mL)的溶液中加入1N水性NaOH(30mL)并且混合物搅拌3小时。混合物经浓缩以去除THF,用1N水性HCl调整至pH8,然后冻干以得到黄色固体的粗制酸7C(5.25g,100%)(包含NaCl),其无须任何纯化可用于下一步骤。1HNMR(400MHz,MeOD)δ3.68(m,4H),2.91(m,4H),2.57(s,3H).步骤3.在0℃下,向内含化合物7C(450mg,2mmol)的DMF(15mL)和DCM(5mL)溶液中加入EDC(400mg,2mmol)和HOBt(310mg,2mol)。在0℃搅拌该混合物30分钟。在0℃下添加化合物1D(500mg,2mmol)。将混合物在40℃下搅拌过夜。混合物用H2O稀释并用EtOAc萃取。有机层在Na2SO4上干燥并浓缩以得到粗产物,其通过柱色谱(石油醚/DCM/MeOH50/50/0至0/10/1)纯化和重结晶(MeOH)以得到暗黄色固体的实施例7(230g,15%,2批)。1HNMR:(400MHz,DMSO-d6).10.74(s,1H),8.61(d,J=8.0Hz,1H),7.56(d,J=8.0Hz,1H),7.30(d,J=8.0Hz,1H),7.09(s,1H),7.05-7.02(m,1H),6.96-6.93(m,1H),4.14(s,1H),3.50(s,4H),2.99-2.84(m,2H),2.42(s,4H),2.21(s,3H),1.57(s,2H),1.25-1.21(m,4H),0.80(s,3H).实施例8:N-(1-(1H-吲哚-3-基)己-2-基)-5-(4-甲基哌嗪-1-基)-4H-1,2,4-三唑-3-羧酰胺.步骤1.在0℃下向内含化合物8A(4.5g,35mmol)的1M硫酸(70mL,70mmol)的搅拌混合物中加入硝酸钠(2.41g,52.5mmol)的水(20mL)溶液,之后加入额外的水(35mL)。25分钟后,加入内含KBr(8.33g,70mmol)和溴化亚铜(4.51g,10.5mmol)的水(35mL)溶液。在20℃下搅拌所得的混合物3小时,并用乙酸乙酯萃取混合物(3x)并且合并的萃取物用盐水(30mL)洗涤、在Na2SO4上干燥,并浓缩至干以得到白色固体的化合物8B(2.9g,43%)。实际ES-API:191.9,189.9步骤2.向内含化合物8B(2.9g,15mmol)的DCM(5mL)和THF(15mL)混合物的溶液中加入Mukaiyama试剂(5g,19.5mmol)和DIPEA(5.8mL)。在搅拌10分钟后,向混合物中加入1-(1H-吲哚-3-基)己-2-胺1D(3.3g,15mmol)。将混合物在50℃下搅拌3小时。用DCM稀释混合物,用水洗涤。有机相在Na2SO4上干燥并浓缩,并且通过在硅胶(20∶1DCM/MeOH)上的柱色谱纯化残留物以得到淡黄色固体的化合物8C(2.7g,47%)。1HNMR(400MHz,CDCl3)δ7.96(brs,1H),7.54(d,J=8.0Hz,1H),7.23(d,J=8.0Hz,1H),7.07-7.11(m,2H),7.01-7.04(m,1H),6.96(brs,1H),4.38(d,J=4Hz,1H),2.95(d,J=4Hz,1H),1.61(brs,1H),1.39-1.46(m,1H),1.19-1.28(m,4H),0.78(s,3H).步骤3.在120℃下,化合物8C(778mg,2mmol)和N-甲基哌嗪(1g,10mmol)的混合物在密封管中过夜搅拌。加入乙酸乙酯(20mL)并滤去沉淀的固体。用水和盐水洗涤滤液。溶液在Na2SO4上干燥,浓缩,并通过制备型TLC(10:1DCM/MeOH)纯化以得到黄色固体的实施例8(184mg,18%)。1HNMR(400MHz,CDCl3)δ8.43(brs,1H),7.54(d,J=8.0Hz,1H),7.26(d,J=8.0Hz,1H),7.19(s,1H),7.05(t,J=8.0Hz,1H),6.96-7.00(m,3H),4.32(d,J=4Hz,1H),3.42(s,4H),2.28-2.29(m,2H),2.50(s,4H),2.28(s,3H),1.56-1.62(m,1H),1.41-1.46(m,1H),1.22-1.33(m,4H),0.79(t,J=8.0Hz,3H).实施例9:N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)-1H-咪唑-5-羧酰胺.步骤1.在0℃下向内含咪唑9A(20g,294mmol)的THF(200mL)和TEA(40g,400mmol)溶液中缓慢加入二甲基氨基磺酰氯(55g,383mmol)。反应混合物在室温下搅拌过夜。将反应混合物倾倒入300mL水中,用乙酸乙酯萃取(3x)。溶液用水和盐水洗涤,在Na2SO4上干燥,然后浓缩至干以得到无色油状的化合物9B(42g,89%),其在置于室温下1小时后固化。该固体物质无需进一步纯化即可直接用于下一步骤。1HNMR(400MHz,CDCl3)δ7.86(s,1H),7.23(s,1H),7.11(s,1H),2.82(s,6H).步骤2.在-78℃下向含化合物9B(10g,57mmol)的无水THF(100mL)溶液中滴加正丁基锂(27.3mL,68mmol)。溶液在该温度下搅拌30分钟。然后在-78℃下加入溴甲烷(20.5g,62.7mmol)并且允许混合物在3小时的时间段内升温置室温,之后在室温(25℃)下过夜连续搅拌。混合物用饱和水性NH4Cl(50mL)稀释并用DCM(3x)萃取。合并的有机层用盐水洗涤,在Na2SO4上干燥并浓缩,并且通过MPLC(己烷/DCM1∶1)纯化以得到轻油状的化合物9C(8.4g,57%)。1HNMR(400MHz,CDCl3)δ7.41(s,1H),7.00(s,1H),3.01(s,6H).步骤3.向内含化合物9C(10g,39mmol)和N-甲基哌嗪(12g,118mmol)的二噁烷(100mL)混合物在90℃下过夜搅拌。将该溶液倾倒入水中,用DCM萃取(3x)。合并的有机层在Na2SO4上干燥并浓缩,并且通过柱色谱(10∶1DCM/MeOH10∶1)纯化残留物以得到棕色固体的化合物9D(3.6g,34%)。1HNMR(400MHz,CDCl3)δ7.27(s,1H),6.84(s,1H),3.43(m,8H),2.97(s,3H),2.91(s,6H).步骤4.在-78℃下向含化合物9D(3.6g,13mmol)的无水THF(40mL)溶液中滴加正丁基锂(6.3mL,16mmol)。溶液在该温度下搅拌30分钟,然后加入氯甲酸甲酯(1.48g,16mmol)。在-78℃下搅拌该混合物3小时,然后升温至室温。混合物用饱和水性NH4Cl(50mL)稀释并用DCM(3x)萃取。合并的有机层在Na2SO4上干燥并浓缩,并且通过柱色谱(60∶1DCM/MeOH)纯化残留物以得到棕色粘稠油状的化合物9E(1.5g,54%)。1HNMR(400MHz,CDCl3)δ7.29(s,1H),3.76(s,3H),3.50(m,4H),2.77(s,6H),2.51(m,4H),2.28(s,3H).步骤5.将化合物9E(1.5g,4.5mmol)和浓HCl(7.5mL)的混合物在60℃下搅拌过夜。混合物真空浓缩并且用MeOH处理残留物(10mL)。通过过滤收集沉淀的白色固体并且真空干燥以得到盐酸盐形式的化合物9F(800mg,84%)。1HNMR(400MHz,D2O)δ7.53(s,1H),4.09(d,J=14Hz,2H),3.67(t,J=12.4Hz,2H),3.59(d,J=12.4Hz,2H),3.31(t,J=12.4Hz,2H),2.97(s,3H).步骤6.向内含化合物9F(1.05g,5mmol)的DCM(2mL)和THF(10mL)溶液中加入Mukaiyama试剂(1.65g,6.5mmol)和DIPEA(1.9g,20mmol)。在搅拌10分钟后,向混合物中加入1-(1H-吲哚-3-基)己-2-胺1D(1.1g,5mmol)并且混合物在50℃下搅拌3小时。真空下去除溶剂并且用乙酸抑制稀释残留物并用水和盐水洗涤,在Na2SO4上干燥,并浓缩。残留物通过制备型TLC(10∶1DCM/MeOH)纯化2次以得到淡黄色固体的实施例9(200mg,8.3%)。1HNMR(400MHz,MeOD-d4)δ7.59(d,J=8Hz,1H),7.30(d,J=8Hz,1H),7.29(s,1H),7.08-7.04(m,1H),7.07(s,1H),6.98-6.94(m,1H),4.33-4.26(m,1H),3.40-3.35(m,4H),2.99-2.97(m,2H),2.70-2.68(m,4H),2.44(s,3H),1.68-1.60(m,1H),1.55-1.45(m,1H)1.38-1.28(m,4H),0.86(t,J=7Hz,3H).实施例10:N-(1-(5-氟-1H-吲哚-3-基)己-2-基)-2-(2-吗啉代乙基)噻唑-5-羧酰胺.步骤1.内含化合物4C(12.7g,44.8mmol)的乙腈(127mL)溶液用吗啉(5.6g,65mmol)和Cs2CO3(21.3g,65mmol)处理并在50℃下过夜搅拌。过滤混合物,浓缩滤液,并且通过柱层析(CH2Cl2/MeOH=10∶1)纯化残留物以得到淡黄色油状的化合物10A(5.6g,63%)。1HNMR(400MHz,MeOD)δ7.66(d,J=3.2Hz,1H),7.19(d,J=3.6Hz,1H),3.72(m,4H),3.22(m,2H),2.77(m,2H),2.52(m,4H).步骤2.在-78℃下向含化合物10A(5.6g,28mmol)的无水THF(56mL)溶液中滴加正丁基锂(13.6mL,17mmol)。混合物在该温度下搅拌30分钟,然后在-78℃下向溶液中滴加氯甲酸甲酯(3.2g,17mmol)。将混合物搅拌3小时。在此期间,温度升温至室温(25℃)。加入饱和NH4Cl水溶液(50mL)以猝灭反应。混合物用EtOAc萃取,用盐水洗涤,在硫酸钠上干燥并浓缩以得到化合物10B(4.0g,60%)。该粗物质无需进一步纯化即可直接用于下一步骤。1HNMR(400MHz,CDCl3)8.27(s,1H),3.90(s,3H),3.72-3.76(m,4H),3.21(t,J=8.0Hz,2H),2.77(t,J=8.0Hz,2H),2.54(s,4H).步骤3.向内含化合物10B(3g,11.7mmol)的MeOH(30mL)溶液中添加内含NaOH(0.7g,17mmol)的水(9mL)溶液。将混合物在25℃下搅拌3小时。真空去除MeOH并且用EtOAc(30mL)洗涤溶液。用1NHCl将水相调整到pH=5-6并且用EtOAc萃取(50mLx3)。有机相用盐水洗涤,在硫酸钠上干燥并浓缩至干以得到橙色固体的化合物10C(3g,100%)。该粗物质无需进一步纯化即可直接用于下一步骤。步骤4.向含化合物10D(3.5g,18mmol)的CH2Cl2(35mL)溶液中加入CDI(3.52g,21.8mmol)。混合物在室温下搅拌2小时,然后向混合物中加入N,O-二甲基盐酸羟胺(2.3g,26mmol)。将混合物在室温下搅拌4小时。混合物用水(30mL)稀释,并用EtOAc(50mLx2)萃取。有机相用盐水洗涤,在硫酸钠上干燥,浓缩以得到棕色油状的化合物10E(4.5g,100%)。1HNMR(400MHz,CDCl3)8.33(s,1H),7.15-7.30(m,3H),6.90-6.92(m,1H),3.87(s,2H),3.73(s,3H),3.28(s,3H).步骤5.在-78℃下向含化合物10E(15mmol,57mmol)的无水THF(72mL)溶液中滴加正丁基锂(46mL,91mmol)。混合物在该温度下搅拌10分钟。加入水性HCl(1M,30mL)以猝灭反应。混合物用EtOAc萃取,用盐水洗涤,在硫酸钠上干燥并浓缩以得到化合物10F(3.5g,70%)。该粗物质无需进一步纯化即可直接用于下一步骤。步骤6.向含AcONH4(46g,0.6mol)和NaBH3CN(9.5g,0.15mol)的MeOH(140mL)和THF(30mL)溶液中加入化合物10F(3.5g,15mmol)。将混合物在室温下搅拌20小时。真空去除MeOH和THF并且向残留物中加入饱和NaHCO3。使用EtOAc萃取溶液(100mLx2)。有机相用盐水洗涤,在硫酸钠上干燥,并浓缩以得到棕色油状的化合物10G(4.0g,100%)。该粗物质无需进一步纯化即可直接用于下一步骤。步骤7.向内含化合物10C(1.6g,6.6mmol)的二氯甲烷(6.4mL)和THF(16mL)溶液中加入Mukaiyama试剂(2.2g,8.6mmo1)和DIPEA(1.6g,6.6mmol)。将所得混合物搅拌10分钟。加入化合物10G(1.6g,6.8mmol)并且混合物在40℃搅拌2小时。滤去所得沉淀并真空浓缩滤液。残留物溶于乙酸乙酯(30mL)中,用水(10mL)和盐水(10mL)洗涤,用硫酸钠干燥,并浓缩。通过制备型HPLC(ShimadzuLC-8A制备型HPLC,Luna(2)C18柱,10mM水性NH4OH中25%-55%CH3CN,在20分钟内,以80mL/分钟)纯化残留物以得到白色固体的实施例10(300mg,9.8%)。1HNMR(400MHz,CDCl3)8.38(s,1H),7.74(s,1H),7.16(t,J=4.4Hz,1H),7.01(s,1H),6.81-6.86(m,1H),5.80(d,J=8.8Hz,1H),4.33(d,J=5.2Hz,1H),3.63(s,4H),3.05(t,J=6.4Hz,2H),2.89-2.94(m,2H),2.64-2.66(m,2H),2.43(s,4H),1.55-1.58(m,1H),1.42-1.44(m,1H),1.22-1.31(m,4H),0.80(t,J=6.8Hz,3H).实施例11:N-(1-(1H-吲哚-3-基)己-2-基)-2-吗啉代噻唑-5-羧酰胺.向内含化合物3A(200mg,0.49mmol)和DIPEA(171μL,0.98mmol)的THF(2mL)混合物中加入吗啉(42μL,0.49mmol)。反应混合物在170℃下在密封的Q-管压力反应器中搅拌1.5小时。加入额外的吗啉(42μL,0.49mmol)并且混合物在170℃下在密封的Q-管压力反应器中加热0.5小时。混合物经浓缩并且通过柱层析(0-5%MeOH/DCM)纯化残留物以得到化合物实施例11(148mg,73%)。1HNMR(400MHz,CDCl3)δ8.08(brs,1H),7.64(d,J=7.5Hz,1H),7.39(s,1H),7.37(d,J=8Hz,1H),7.19(dd,J=7.5Hz,1H),7.12(dd,J=7.5Hz,1H),7.05(d,J=2Hz,1H),5.57(d,J=8Hz,1H),4.39-4.42(m,1H),3.80(t,J=4.5Hz,4H),3.51(t,J=4.5Hz,4H),3.06(dd,J=14.5,5.5Hz,1H),3.01(dd,J=14.5,5.5Hz,1H),1.26-1.64(m,6H),0.87(t,J=7Hz,3H).ESMS+:413.6[M+1].可按照上述方法制备实施例12-26。实施例27:(S)-N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺,和实施例28:(R)-N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺.使用ChiralpakAD-H柱(250x30mm;5μM同上)在THAR80制备型SFC上分离对映异构体。对映异构体溶于甲醇(50mg/mL)中并且每次注射加载45mg的对映异构体。使用70g/流速的CO2中40%2-丙醇(添加剂:0.05%NH3H2O)的流动相和100巴的系统背压来实现分离。柱温度保持在40℃下并且在220nm处检测峰。总循环时间是6分钟。实施例27:((S)-N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺):LCMS(XtimateC18,2.1X30mm,3μM同上):峰1RT:2.036(100%)MS:426.2;任选旋转(DichromPolaraizer,589nM)-0.143(sd=0.0004);手性纯度检验(OJ-H,40%MeOH(0.05%DEA))峰1RT:3.61(99.87%),峰2RT:5.3(0.13%)。实施例28:((R)-N-(1-(1H-吲哚-3-基)己-2-基)-2-(4-甲基哌嗪-1-基)噻唑-5-羧酰胺):LCMS(XtimateC18,2.1X30mm,3μM同上):峰1RT:2.035(98.77%)MS:426.2.峰2RT:2.373(1.23%)MS:427.2;任选旋转(DichromPolaraizer,589nM)+0.160(sd=0.0003);手性纯度检验(OJ-H,40%MeOH(0.05%DEA))峰1RT:3.6(0.18%),峰2RT:5.23(99.82%)。生物实施例1:用α-突触核蛋白肽片段(4F)的体外荧光偏振试验。荧光偏振试验测试了化合物抑制α-突触核蛋白肽片段的自聚集的能力。在测试化合物存在或不存在的情况下(化合物浓度为33.3至0.3μM),肽在室温下孵育60分钟。使用485nm处的激发和520nm处的发射,在荧光偏振模式下的BMGPherastar酶标仪上读取样品。使用四参数逻辑拟合来分析数据(XLFit,IDBS软件)。由美国肽公司(AmericanPeptide)制备肽4F(CTGFVKKDQLGK(SEQIDNO:1))。新鲜肽样品以5mM在纯化水中重建并且稀释到含50mMNaCl的pH7.4的50mMHEPES中至100nM的终浓度。固体化合物溶于DMSO(10mM)中,然后在缓冲剂中稀释。测试的化合物的数据示于表1。也测试了比较化合物A和B。比较物A:比较物B:表1.实施例IC50(μM)*比较物A>30§比较物B6.3513.33±1.9¥23.5530.2745.951.360.3970.3780.7798.5270.4±0.3§280.7±0.4§*n=1除非另外说明§n=2¥n=3±SEM生物实施例2:体内药代动力学试验本文所述化合物的药代动力学和脑部分布在单次静脉内或口服给药后的雄性C57BL/6小鼠中确定。一组54只雄性小鼠被分成两组27只小鼠。以10mg/kg(i.v.)或2mg/kg(p.o.)的测试化合物给予组1(i.v.)和组2(p.o.)中的动物。在给药前和给药后0.08、0.25、0.5、1、2、4、8、和24小时(i.v.),以及给药前和给药后0.25、0.5、1、2、4、6、8和24小时(p.o.)收集血液样品。在各时间点从三只小鼠的组中收集血液到含K2EDTA作为抗凝剂的标记的微量离心管中。通过全血离心分离血浆样品并且在-70℃以下储存直至生物分析。在收集血液样品之后,通过CO2窒息法人道地处死小鼠并且同时收集脑部。收集后,脑部样品在冰冷的磷酸盐缓冲盐水(pH7.4)中洗涤,在滤纸上温和干燥,称重并置于聚丙烯管中。使用磷酸盐缓冲盐水pH7.4匀化其他脑部样品并且总的匀浆体积是脑部重量的三倍。样品然后储存在-70℃以下至生物分析。所有样品经处理用于通过使用乙腈的蛋白质沉淀分析,并且用量身定做的LC/MS/MS方法分析(LLOQ:1.01ng/mL针对血浆和脑部)。使用PhoenixWinNonlin(版本6.3)的非房室分析工具来计算药代动力学参数。从该试验中获得的数据示于表2。表2.*10mg/kg下**2mg/kg下NC=没有检测到化合物(低于检测限)ND=未测定生物实施例3:测试化合物对α-突触核蛋白与脂膜相互作用的影响的NMR试验为了检测脂膜存在下测试化合物与全长ASYN的相互作用,进行了NMR试验。在Varian直接驱动600MHz和VarianInova800MHz光谱仪上在20mM磷酸盐,pH=7.4,100mMNaCl中进行NMR测量,使用10%D2O作为锁定溶剂。使用NMRPipe处理光谱(参见,F.Delaglio,S.Grzesiek,G.W.Vuister,G.Zhu,J.Pfeifer,A.Bax,JBiomolNMR1995,6,277-293)。α-突触核蛋白以0.12mM的浓度使用,同时在存在时以0.8mg/ml加入1-棕榈酰-2-油酰-sn-甘油-3-磷酸甘油(POPG)-脂质体。用SOFAST脉冲序列来剂量所有1H-15N相关光谱(参见P.Schanda,E.Kupce,B.Brutscher,JBiomolNMR2005,33,199-211)。易于从之前的出版物中获得靠近生理条件处的谱峰归属(BMRBID16300;参见J.N.Rao,Y.E.Kim,L.S.Park,T.S.Ulmer,JMolBiol2009,390,516-529)。为了配体滴定,向脂质体/ASYN混合物中逐步加入实施例1。记录各步骤的15N-1H相关光谱并且信号强度参照游离形式的ASYN,同时考虑稀释影响。为了减少获得的数据的噪音,ASYN的多个酰胺位置的强度比相对于对应之前观察到的SL1和SL2结合模式选择的2个区域取平均(参见C.R.Bodner,A.S.Maltsev,C.M.Dobson,A.Bax,Biochemistry2010,49,862-871)。如图1所示,当ASYN包埋在脂膜中时,ASYN的异核单量子相干(HSQC)光谱信号强度衰减。HSQC信号的脂质诱导的衰减被实施例1逆转,因此证明了测试化合物破坏ASYH与脂膜结合的能力。图1A显示在POPG脂质体存在下随着ASYN残基变化的信号衰减。Y轴(I/Io)是在脂膜存在(I)或不存在(Io)下ASYN的HSQC光谱信号强度的比率。在图1B中,ASYN残基3-23的平均I/Io比率以添加的实施例1的浓度的函数作图。该曲线显示实施例1以浓度依赖性方式逆转了ASYN与1-棕榈酰-2-油酰-sn-甘油-3-磷酸甘油(POPG)(0.8mg/mL)脂质体的相互作用。当分析ASYN残基66-76时获得类似结果。生物实施例4:测试化合物对脂膜中环形低聚物的影响使用电子显微镜来直接观察测试化合物对脂膜中ASYN低聚物形成的影响。用50%ETOH中的饱和乙酸铀酰溶液对带脂膜的福尔瓦网格复染25分钟。网格然后在2%碱式硝酸铋的液滴上漂浮10分钟,并再次用双蒸水小心淋洗,并使其完全干燥。使用ZeissEM10透射电子显微镜来对网格进行成像。对于各网格,获得了10000倍放大的5-10个电子显微图和40000倍放大的5-10个图像。扫描最好的底片并用ImageJ1.43程序分析来估计每个高倍视野(100x100nm)中环形低聚物的数量(Rasband,W.S.,ImageJ,美国马里兰州贝塞斯达的国立卫生研究院(U.S.NationalInstitutesofHealth,Bethesda,Maryland,USA),http://imagej.nih.gov/ij/,1997-2014)。在该研究中,发现实施例1显著降低了脂膜中ASYN的环形,环状,低聚形式的积累,同时仍然观察到小的非环形聚集体。图2A显示在实施例1存在或不存在的情况下,在脂质包被的福尔瓦网格上形成的ASYN低聚物的电子显微图像。图2B是反应电子显微图像定量的照片。实施例1将福尔瓦网格上检测到的环形ASYN低聚物的数量降低至10nM的浓度(20次测量的平均值±SEM)。实施例1在相对于ASYN低于化学计量浓度下实现这些效果。这些发现与显示实施例1使ASYN构象稳定化不太可能在脂膜中形成环状低聚物的分子动力学建模相一致。这些结果表明实施例1以降低ASYN低聚物对脂膜的亲和性的方式与ASYN的低聚和脂质结合形式相互作用。实施例1能够干扰ASYN低聚化、ASYN与脂膜的结合、以及在这些膜中形成环形环状低聚物(“孔”)。这些结果表明实施例1改变了ASYN的聚集并且防止被认为导致帕金森病中错误折叠、低聚化的ASYN的神经毒性的特定低聚结构的形成。生物实施例5:测试化合物对细胞中α-突触核蛋白的影响研究了实施例1对过表达人ASYN的B103神经母细胞瘤细胞中ASYN积累的影响。使用慢病毒表达系统来在这些细胞中表达GFP-标记的ASYN。在表达开始后48小时,加入载剂或实施例1(0.1或1.0μM)持续另外24小时。然后观察累积的GFP-ASYN的量。如图3所示,实施例1在1.0μM下降低了这些细胞中GFP荧光的强度(对比载剂对照组*p<0.05)。因此,发现实施例1降低了ASYN-过表达细胞中ASYN-GFP的浓度。生物实施例6:体内功效研究帕金森病(PD)的特征在于α-突触核蛋白(ASYN)的低聚形式的异常累积。假设ASYN的这些毒性形式部分通过在细胞膜中形成孔状结构导致PD和共核蛋白病中观察到的神经功能障碍和细胞死亡。本文所述的化合物设计为通过阻断这些ASYN毒性物质的形成和累积缓解PD-相关症状和致病性。A)帕金森病的转基因小鼠模型在Thy-1启动子下过表达人野生型ASYN的PD转基因小鼠(也称为种系61ASYN转基因小鼠)模型中评价实施例1,通过以每天一次给予0、1、或5mg/kg(i.p.)的实施例1(每周五天)持续3个月,然后评价PD-相关的感觉运动性能、生物化学改变、以及ASYN和相关蛋白质的神经病理变化。使用圆点束任务来评价感觉运动损伤,使用滑跌(slip)的数量作为主要结果测量(图4)。为了确认转基因小鼠模型的活力,测试了ASYN转基因和非转基因小鼠,并且载剂处理的转基因对象的滑跌的数量在统计学上明显高于载剂处理的非转基因对照组(****p<0.0001)。在两个测试剂量下用实施例1处理的转基因小鼠在滑跌上相比载剂处理的转基因小鼠在统计学上显著下降(对比载剂处理的ASYN转基因小鼠,#p<0.05和##p<0.01)。对大脑皮质和海马区脑部匀浆的Western印迹分析显示在转基因ASYN蛋白水平上统计学上显著降低。进行了在皮质匀浆中使用A11抗体点印迹方法对低聚蛋白质(包括SAYN)的生物化学评价。验证了转基因小鼠模型,因为对皮质匀浆中低聚物的A11抗体点印迹评价显示相对于载剂处理的非转基因对照小鼠,载剂处理的ASYN转基因小鼠中额叶皮质的胞质部分在A11免疫染色中的统计学显著增加(图5;*p<0.05)。用实施例1(5mg/kg)处理相对于载剂处理的ASYN转基因小鼠在来自小鼠的额叶皮质区的胞质部分中产生了低聚物的统计学显著降低(图5;###p<0.001)。B)种系61ASYN转基因小鼠模型Masliah及同事之前的免疫标记研究已经证明在种系61ASYN转基因小鼠中的皮质神经毡中ASYN免疫标记的统计学显著增加(MasliahE.等,Science,2000,287(5456):1265-9)。这些神经病理发现在使用Masliah和同事所述的方法的现有研究中被再次确认。实施例1给药(1和5mg/kg剂量)在ASYN水平上产生了统计学上显著的减少,如对ASYN免疫标记的影响所确定(图6和7)。相对于非转基因/载剂对照,在ASYN转基因小鼠的皮质神经毡(****p<0.0001)(图6A)和神经细胞体(**p<0.01)(图6B)中用密理博抗-α-突触核蛋白抗体的ASYN免疫标记在统计学上显著增加。实施例1(1和5mg/kg)给药在皮质神经毡中的α-突触核蛋白免疫标记中产生了统计学显著降低(图6A)(对于载剂处理的ASYN转基因小鼠####p<0.0001),以及在5mg/kg下在ASYN免疫标记的神经元细胞体数量的非统计上显著降低(图6B)。此外,观察到包括酪氨酸羟化酶、NeuN、和GFAP的神经变性相关标志物的中和。如图8所示,使用上述的圆点束表现试验研究了0.5mg/kg和1mg/kg的实施例1对种系61ASYN转基因小鼠中感觉运动损伤的影响。与载剂处理的非转基因对照对象相比,在载剂处理的ASYN转基因对照小鼠中观察到滑跌数量的统计学显著增加(****p<0.0001)。相对于载剂处理的ASYN转基因小鼠,在ASYN转基因小鼠中,在1mg/kg下,实施例1处理产生了统计学显著的改善(减少的滑跌)(##p<0.01)。在0.5mg/kg下,实施例1产生了滑跌的非统计学显著减少。总之,这些结果证明实施例1在转基因小鼠模型中显著改善了感觉运动性、生物化学、和神经病理学结果。这些发现确认给予实施例1在帕金森病/路易体痴呆(PD/DLB)的转基因小鼠小鼠模型中产生的行为、生物化学、和神经病理学测量的改善。生物实施例7:生物标志物的开发A)粪便勃利计数在开发可转化功能性和生物化学生物标志物的努力中,进行了其他评价,包括评价在新环境中产生的粪便勃利计数和ASYN的死后心脏水平。帕金森患者的慢性便秘患病率为50-80%,并且可能在诊断之前出现20年以上(Awad,R.A.WorldJ.Gastroenterol.2011,17(46),5035-5048;Kim,J.S.等,J.Neurol.Sci.2011,310(1-2),144-151)。相关的症状包括降低的通便次数和EMG异常(括约肌,直肠肛门抑制反射),并且伴随着关键的病理学发现,包括副交感神经和神经中的路易体,和降低的多巴胺能神经元。这种带有降低的活性水平的肠功能异常改变了饮食(食物和水),以及帕金森药物的效果。较早出版的研究包括种系61ASYN转基因小鼠有降低的结肠运动、粪便排除、和体重的报告(Wang,L.等,Neurogastroenterol.Motil.2012,24(9),e425-436)。对于本发明的研究而言,粪便勃利计数的评价与自发运动活性测试期间联用。在5分钟测试期间结束时,实验人员对测试室中存在的粪便勃利数量进行计数,并且结果示于图9。相对于载剂处理的非转基因对照小鼠,载剂处理的ASYN转基因小鼠在新环境中产生的粪便勃利在统计学上明显减少(*p<0.05)。实施例1对非转基因小鼠中产生的勃利数量没有影响,但是在0.5mg/kg(#p<0.05对比载剂处理的ASYN转基因小鼠)和1mg/kg(###p<0.001对比载剂处理的ASYN转基因小鼠)下在ASYN转基因小鼠中恢复了功能。B)心脏功能向肠功能障碍那样,心脏生物化学和功能的变化可能出现在帕金森诊断前20年或更早。PD患者中充分表征的功能改变包括改变的心律差异和体位性低血压(Kaufmann,H.等,HandbookClin.Neurol.2013,117,259-278;Jain,S.等,Neurobiol.Dis.2012,46(3),572-580;Senard,J.M.等,Rev.Neurol.(Paris)2010,166(10),779-784;Post,K.K.等,ParkinsonismRelat.Disord.2008,14(7),524-531)。这些功能变化伴随着心肌去甲肾上腺素能神经丢失的病理学发现和在心脏自主神经中存在ASYN聚集(Jellinger,K.A.,J.Neurol.Sci.2011,310(1-2),107-111)。在种系61ASYN转基因小鼠中之前的心脏功能和生物化学表征已经证明位于去甲肾上腺素能神经内的心脏的心室和心房壁中存在hASYN(Fleming,S.M.,J.ParkinsonsDis.2011,1(4),321-327)。对于目前的研究,通过Western印迹分析进行对心脏ASYN的死后Western印迹评价以确认在ASYN转基因心脏组织中的存在并且在ASYN的转基因心脏水平上评价实施例1的影响(图10)。相对于载剂处理的非转基因对照小鼠,在载剂处理的ASYN转基因小鼠中在检测的ASYN心脏水平上有统计学显著增加(***p<0.001)。相对于载剂处理的ASYN转基因小鼠,用0.5或1mg/kg的实施例1处理的ASYN转基因小鼠中ASYN水平有统计学显著的正常化(####p<0.0001)。C)视网膜成像已经假设神经蛋白α-突触核蛋白(ASYN)的异常聚集是帕金森病(PD)和路易体痴呆(DLB)中神经细胞死亡和突触功能失调的原因。已经开发了选择性干扰α-突触核蛋白折叠动力学并防止转播二聚体形成的化合物并在动物模型中进一步评价。在一些帕金森患者中存在视觉功能改变(Botha,H.等,ParkinsonismRelat.Disord.2012,18(6),742-747;Bodis-Wollner,I.等,Behav.Neurosci.2013,127(2),139-150;Bodis-Wollner,I.,ParkinsonismRelat.Disord.2013,19(1),1-14;Javaid,M.A.等,ParkinsonismRelat.Disord.2012,18(补充1),S100-3)并且最近的报告已经表明PD视网膜中的潜在病理学变化。光学相干断层成像术(OCT)研究已经证明帕金森患者中视网膜神经毡层减少(Yu,J.G.等,PLoSOne2014,9(1),e85718)。死后评价已经显示PD视网膜中ASYN沉积(Bodis-Wollner,I.等,Ann.Neurol.2014,75(6),964-6)。之前证明了在DLB/PD的PDNG78转基因小鼠模型中e-GFP-ASYN的报告纵向视网膜成像评价作为评价和跟踪帕金森病的动物模型中神经变性变化进展的方法的可行性(Rockenstein等,“PD/DLB的转基因小鼠模型中α-突触核蛋白-eGFP的视网膜扫描评价(Retinalscanningevaluationsofalpha-synuclein-eGFPdepositioninatransgenicmousemodelofPD/DLB),”神经科学协会(SocietyforNeurosciences),年会,2013,摘要329.06)。PDNG78视网膜中进行性病理学特征显示反映CNS病理学,从而提供了非侵入性和重复评价PD/DLB的转基因小鼠模型中潜在治疗介入的手段。在帕金森病/路易提痴呆(PD/DLB)的转基因小鼠模型中的纵向视网膜成像研究中,进行该研究以确定实施例1(3个月,0和5mg/kg下i.p.给药)对α-突触核蛋白(ASYN)视网膜病理学的存在和进展的影响。转基因小鼠对象过表达PDGF-β启动子下的融合α-突触核蛋白-GFP(绿色荧光蛋白)并且通常称为PDNG78转基因小鼠(Rockenstein,E.等,J.Neurosci.Res.2005,80,247-259)。PDNG78转基因小鼠以比非转基因对照小鼠高2-5倍的水平表达融合的ASYN-GFP。ASYN的CNS表达水平在包括新皮层和海马区的PDNG78转基因小鼠的边缘系统中最高。ASYN-GFP的细胞分布反映了共核蛋白病相关的特征,包括神经细胞体中的积累、神经毡的弥漫染色、突触点状染色、和血管周围沉积。进行了总共四个成像期间,包括在开始处理之前的基线,和以大约1个月间隔的3次后续成像期间。对视网膜图像中含ASYN-GFP的图像的百分比的分析(图11)显示在开始处理之前,在基线处的转基因小鼠的视网膜中含ASYN-GFP的图像面积百分比(**p<0.01对比载剂处理的非转基因小鼠)和对于各后簇扫描(*p<0.05和***p<0.001对比载剂处理的非转基因小鼠)。在处理约60天后,在用实施例1处理(5mg/kg)的转基因小鼠中ASYN-GFP阳性面积百分比降低,并且持续至90天成像时间点(###p<0.001对比载剂处理的ASYN转基因小鼠)。对ASYN-GFP阳性颗粒计数的分析(图12)显示在转基因小鼠中增加和持续的血管周围和神经末端绿色荧光蛋白(GFP)标记,但在非转基因小鼠中没有显示。在开始处理前的基线处(*p<0.05对比载剂处理的非转基因小鼠)和在处理约60天时开始的扫描(**p<0.01对比载剂处理的非转基因小鼠),转基因小鼠的视网膜中的总ASYN-GFP颗粒计数的统计学显著增加。在处理约60天后,在用实施例1处理(5mg/kg)的ASYN转基因小鼠中ASYN-GFP阳性颗粒数量降低,并且持续至90天成像时间点(##p<0.07对比载剂处理的ASYN转基因小鼠)。来自该研究的发现证明给予实施例1(每天5mg/kg;持续3个月)在作为PD/DLB模型的过表达ASYN的转基因小鼠的ASYN视网膜病理学中产生了有益的变化。这些数据也通过潜在可转化的成像方法提供了对实施例1的有益影响的其他证据测量。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1