具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物的制作方法
本发明涉及一种亲水性聚合物衍生物,该亲水性聚合物衍生物具有能够酸水解的缩醛连接基团,并且用于生物官能性分子的化学改性,所述生物官能性分子例如有生理活性的蛋白质、肽、抗体、核酸或低分子量药物以及例如脂质体和聚合物胶束的药物载体。
背景技术:
在药物递送系统中,使用具有低抗原性的亲水性聚合物对生物官能性分子或药物载体的化学改性是提高这些药物等的水溶解度和生物利用度以及延长血液中的循环时间的有效技术。另一方面,已知在结合于亲水性聚合物的药物等被运送至将为目标的组织或位点后,由亲水性聚合物形成的水合层降低与细胞膜的相互作用,并且抑制例如向细胞内的摄入和核内体逸出的体内/细胞内动力学。对于此问题,已经进行了一种通过在适当的时间将亲水性聚合物链从药物等脱附来克服问题的方法。大多数策略利用了生命体各个部分的环境变化,例如,还原性环境或者特定酶的存在或缺失,作为亲水性聚合物链的脱附的触发,并且其中之一是利用pH变化的技术。
已知与正常组织相比,生命体中肿瘤组织的周边为酸性环境,并且在将药物等通过内吞通道引入细胞内后,核内体内部的pH也逐渐降低。因此,出于在酸性环境下选择性脱附亲水性聚合物的目的,已经报道了大量的具有引入其结构中的酸可水解缩醛连接基团的亲水性聚合物衍生物的合成实例。然而,没有其中能够控制缩醛连接基团的水解性的实例,并且不少实例在引入缩醛连接基团的方法中存在问题。
例如,专利文献1公开了一种分枝型聚乙二醇衍生物及其合成方法,其中作为具有低抗原性的水溶性聚合物的两条聚乙二醇链通过衍生自各种醛或酮的缩醛基连接,但是并未描述水解性的任何评估数据。而且,文中描述的合成方法是通过将过量的聚乙二醇与各种醛或酮反应,以获得具有缩醛连接基团的聚乙二醇衍生物的方法,从而在反应后残留了大量未反应的聚乙二醇。在使用混合物作为原材料进行聚合物末端激活的情况下,形成了其中未反应的聚乙二醇的末端也被激活的作为副产物的杂质。当将包含这样的杂质的激活的聚乙二醇用于药物改性时,结果形成了用不包含缩醛连接基团的聚乙二醇改性的药物,从而在药物的体内动力学和物理性质上产生的很大影响。因此,需要在与药物反应前去除聚乙二醇杂质,但是在工业规模生产的情况下,从技术和成本的观点来看,有着聚合物杂质的分离和去除导致严重不良效果的可能性。
获得缩醛化合物的另一种方法是将醇与乙烯基醚在酸性条件下反应的方法。例如,在非专利文献1中,通过将具有各种官能团的乙烯基醚与聚乙二醇反应,合成了具有通过亚乙基缩醛连接基团连接的各种官能团的聚乙二醇衍生物。然而,在此文献中,也没有给出水解性的评估数据。
在非专利文献1中描述的合成方法中,由于作为低分子量化合物的乙烯基醚相比于聚乙二醇过量使用,所以没有残留大量的未反应的聚乙二醇。然而,通过该合成方法引入的缩醛基包含亚乙基缩醛结构,因此能够引入的缩醛基的类型受到限制。在引入苯亚甲基缩醛基的情况下,不可避免地形成缩酮结构,并且由于缩酮结构对酸敏感,在合成方法中,通过缩酮交换反应大量形成其中两条聚乙二醇链通过缩酮基团连接的作为副产物的二聚体杂质。因此,非专利文献1中描述的合成方法难以应用于具有苯亚甲基缩醛连接基团的聚乙二醇衍生物的合成。
另一方面,在非专利文献2中,通过合成具有通过使用低分子量模式药物的羟基而形成的缩醛基的单元并且将单独合成的激活的聚乙二醇衍生物与该单元缩合,合成了其中低分子量模式药物通过脂肪族或苯亚甲基缩醛连接基团连接的几种类型聚乙二醇衍生物。在此情况中,尽管已经示出了缩醛基周边的结构差异影响水解速率,即聚乙二醇链的脱附速率,但是并未阐明该速率与缩醛基周边的结构的相关性,并且因此不能认为能够控制水解性。同样地,由于该方法是使用低分子量模式药物的羟基形成缩醛基的方法,所以难以在除低分子量药物以外的物质、例如蛋白质和药物载体等的化学改性中使用此方法。
如上所述,尽管有出于在生命体中的酸性环境下脱附亲水性聚合物链的目的、各自具有引入结构中的缩醛连接基团的亲水性聚合物衍生物的许多实例,但是没有关于能够在任意pH下准确控制其中缩醛连接基团的水解速率、即亲水性聚合物链的脱附速率的亲水性聚合物衍生物的实例。
现有技术文献
专利文献
专利文献1:WO2005/108463
非专利文献
非专利文献1:聚合物科学会第57届年度会议,日本,Preprints of The Society of Polymer Science,日本,57,1897(2008年5月)
非专利文献2:Bioconjugate Chem.2004,15,1254-1263
发明概述
本发明待解决的问题
在生命体的各个部分中pH的偏差非常小,并且例如尽管肿瘤组织的外周与正常组织中7.4的pH相比是酸性环境,其pH为弱酸性,大约为6.0。同样地,核内体内部显示出5.5至6.0的pH并且是弱酸性,并且核内体内部逐渐地酸化并且接近溶酶体的pH,即4.5至5.0的pH。由于核内体最终与溶酶体融合,需要的是,为了避免其被溶酶体酶降解,摄入核内体内的药物等应该在约5.5的pH从核内体逸出。因此,在意欲通过使用在例如肿瘤组织的外周或核内体内部的生命体的各个部分的pH中的微小区别而脱附亲水性聚合物链而控制例如位点选择的细胞摄入和药物等的核内体逸出的体内/细胞内动力学的情况下,需要准确地控制在生命体中的弱酸性环境的pH下,缩醛连接基团的水解速率。
本发明的目的是提供一种亲水性聚合物衍生物,该亲水性聚合物衍生物具有能够准确控制其在生命体中的弱酸性环境的pH下的水解速率的缩醛连接基团,并且其在水解时不释放出亲水性聚合物链或连接的药物等以外的低分子量物质,更具体为低分子量醛类。
解决问题的手段
为了解决上述问题而进行了大量研究,作为结果,本发明人开发出一种亲水性聚合物衍生物,该亲水性聚合物衍生物具有能够准确控制其在生命体中的弱酸性环境的pH下的水解速率的环状苯亚甲基缩醛连接基团,并且其在水解时不释放出亲水性聚合物链或连接的药物等以外的低分子量物质,更具体为低分子量芳香醛类。
本发明的特征在于,化学反应性官能团与亲水性聚合物通过具有取代基的环状苯亚甲基缩醛连接基团连接。通过适当地选择环状苯亚甲基缩醛连接基团的苯环上取代基的类型和位置,能够调节影响缩醛连接基团的水解速率的缩醛基周边的电子密度和立体位阻的程度。基于此特征,可以给予缩醛连接基团理想的水解速率,并且从而可以以任意速率将亲水性聚合物链从连接至亲水性聚合物衍生物的药物等脱附。
本发明的另一个特征在于,亲水性聚合物和环状苯亚甲基缩醛连接基团的苯环通过在生命体中稳定的键连接。基于此特征,本发明具有这一优点,即在水解时能够避免释放出亲水性聚合物链或连接的药物等以外的低分子量物质,更具体为低分子量芳香醛类。
本发明的亲水性聚合物衍生物能够通过执行其中引入具有取代基的环状苯亚甲基缩醛基的连接基团化合物和亲水性聚合物中间体之间的偶联反应而合成。因此,不需要如专利文献1中这样使用过量的亲水性聚合物以形成缩醛基,并且不需要去除未反应的亲水性聚合物杂质,从而容易以工业规模进行生产。另外,对能够引入的缩醛基的类型没有限制,并且能够引入不能通过非专利文献1的合成方法引入的苯亚甲基缩醛基。此外,不同于非专利文献2,在本发明的亲水性聚合物衍生物中,由于能够在缩醛连接基团末端引入各种官能团,所以可能与各种生物官能性分子和药物载体形成共价键。
即,本发明包括下列各项。
[1]一种亲水性聚合物衍生物,该亲水性聚合物衍生物具有环状苯亚甲基缩醛连接基团,由下列式(1)表示:
其中R1和R6各自独立地是氢原子或烃基;R2、R3、R4和R5各自独立地是吸电子或给电子取代基或氢原子;X1是化学反应性官能团;P是亲水性聚合物;s是1或2,t是0或1,并且s+t是1或2;w是1至8的整数;并且Z1和Z2是各自独立地选择的二价间隔物。
[2][1]的亲水性聚合物衍生物,其中s是1并且t是0,R2和R5各自独立地是氢原子,并且R3、R4和P-Z1的取代基常数(σ)的总和(Σσ)满足-0.30≤Σσ≤1.05。
[3][1]的亲水性聚合物衍生物,其中s是1并且t是0,R2和R5中的至少一个是上述取代基,并且R3、R4和P-Z1的取代基常数(σ)的总和(Σσ)满足-1.71≤Σσ≤0.88。
[4][1]的亲水性聚合物衍生物,其中s是1并且t是1,或者s是2并且t是0,R2和R5各自独立地是氢原子,并且R3、R4和P-Z1的取代基常数(σ)的总和(Σσ)满足-0.19≤Σσ≤0.57。
[5][1]的亲水性聚合物衍生物,其中s是1并且t是1,或者s是2并且t是0,R2和R5中的至少一个是上述取代基,并且R3、R4和P-Z1的取代基常数(σ)的总和(Σσ)满足-0.98≤Σσ≤0.48。
[6][1]至[5]任意一项的亲水性聚合物衍生物,其中X1选自由活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基和叠氮基所组成的组。
[7][1]至[6]任意一项的亲水性聚合物衍生物,其中X1选自由式(a)、式(b)、式(c)、式(d)、式(e)、式(f)、式(g)、式(h)、式(i)、式(j)、式(k)、式(l)、式(m)和式(n)所组成的组:
其中R7是氢原子或磺基;R8和R11各自独立地是氢原子或具有1至5个碳原子的烃基;R9是具有1至10个碳原子的烃基,该烃基可以包含卤素原子;并且R10是选自氯原子、溴原子和碘原子的卤素原子。
[8][1]至[7]任意一项的亲水性聚合物衍生物,其中Z1和Z2各自独立地是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基,并且在Z1和Z2的至少一个是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,该结构单元的数量为2以下。
[9][1]至[8]任意一项的亲水性聚合物衍生物,其中P是在末端具有烃基或化学反应性官能团的直线型聚乙二醇。
[10][9]的亲水性聚合物衍生物,其中w是1并且P由式(2)表示:
Y-(OCH2-CH2)n- (2)
其中Y是具有1至24个碳原子的烃基;并且n是3至2000的整数。
[11][9]的亲水性聚合物衍生物,其中w是1并且P由式(3)表示:
X2-Z3-(OCH2CH2)n- (3)
其中X2是不同于X1的化学反应性官能团;Z3是二价间隔物;并且n是3至2000的整数。
[12][1]至[8]任意一项的亲水性聚合物衍生物,其中P是在末端具有不同于X1的烃基或化学反应性官能团的支链型聚乙二醇。
[13][12]的亲水性聚合物衍生物,其中w是1并且P由式(4)表示:
其中Y是具有1至24个碳原子的烃基;n是3至1000的整数;并且v是0或2。
[14][12]的亲水性聚合物衍生物,其中w是1并且P由式(5)表示:
其中X2是不同于X1的化学反应性官能团;Z3是二价间隔物;n是3至1000的整数;并且v是0或2。
[15][12]的亲水性聚合物衍生物,其中w是v+2并且P由式(6)表示:
其中X2是不同于X1的化学反应性官能团;Z3是二价间隔物;n是3至1000的整数;并且v是0或2。
[16][11]、[14]和[15]任意一项的亲水性聚合物衍生物,其中X2选自由活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基和叠氮基所组成的组。
[17][11]、[14]和[15]任意一项的亲水性聚合物衍生物,其中X2选自由式(a)、式(b)、式(c)、式(d)、式(e)、式(f)、式(g)、式(h)、式(i)、式(j)、式(k)、式(l)、式(m)和式(n)所组成的组:
其中R7是氢原子或磺基;R8和R11各自独立地是氢原子或具有1至5个碳原子的烃基;R9是具有1至10个碳原子的烃基,该烃基可以包含卤素原子;并且R10是选自氯原子、溴原子和碘原子的卤素原子。
[18][11]、[14]和[15]任意一项的亲水性聚合物衍生物,其中Z3是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基,并且在Z3是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,该结构单元的数量为2以下。
[19][1]至[8]任意一项的亲水性聚合物衍生物,其中P是具有2至8的末端数的聚乙二醇,构成P的聚乙二醇全部末端各自连接至Z1,并且w等于聚乙二醇的末端数。
[20][19]的亲水性聚合物衍生物,其中P选自由式(r)、式(s)、式(t)、式(u)和式(v)所组成的组:
其中n是3至2000的整数,并且在P由式(r)表示时w是2、在P由式(s)表示时w是3、在P由式(t)表示时w是4、在P由式(u)表示时w是4并且在P由式(v)表示时w是8。
[21]一种由式(55)表示的环状苯亚甲基缩醛连接基团化合物:
其中R1和R6各自独立地是氢原子或烃基;R2、R3、R4和R5各自独立地是吸电子或给电子取代基或氢原子;X3和X4可以相同或不同,各自是化学反应性官能团;s是1或2,t是0或1,并且s+t是1或2;并且Z1和Z2是各自独立地选择的二价间隔物。
[22][21]的环状苯亚甲基缩醛连接基团化合物,其中s是1并且t是0,R2和R5各自独立地是氢原子,并且R3、R4和X3-Z1的取代基常数(σ)的总和(Σσ)满足-0.30≤Σσ≤1.05。
[23][21]的环状苯亚甲基缩醛连接基团化合物,其中s是1并且t是0,R2和R5中的至少一个是上述取代基,并且R3、R4和X3-Z1的取代基常数(σ)的总和(Σσ)满足-1.71≤Σσ≤0.88。
[24][21]的环状苯亚甲基缩醛连接基团化合物,其中s是1并且t是1,或者s是2并且t是0,R2和R5各自独立地是氢原子,并且R3、R4和X3-Z1的取代基常数(σ)的总和(Σσ)满足-0.19≤Σσ≤0.57。
[25][21]的环状苯亚甲基缩醛连接基团化合物,其中s是1并且t是1,或者s是2并且t是0,R2和R5中的至少一个是上述取代基,并且R3、R4和X3-Z1的取代基常数(σ)的总和(Σσ)满足-0.98≤Σσ≤0.48。
[26][21]至[25]任意一项的环状苯亚甲基缩醛连接基团化合物,其中X3和X4各自独立地选自由活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基、叠氮基和羟基所组成的组。
[27][21]至[26]任意一项的环状苯亚甲基缩醛连接基团化合物,其中X3和X4各自选自由式(a)、式(b)、式(c)、式(d)、式(e)、式(f)、式(g)、式(h)、式(i)、式(j)、式(k)、式(l)、式(m)、式(n)和式(o)所组成的组:
其中R7是氢原子或磺基;R8和R11各自独立地是氢原子或具有1至5个碳原子的烃基;R9是具有1至10个碳原子的烃基,该烃基可以包含卤素原子;并且R10是选自氯原子、溴原子和碘原子的卤素原子。
[28][21]至[27]任意一项的环状苯亚甲基缩醛连接基团化合物,其中Z1和Z2各自独立地是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基,并且在Z1和Z2的至少一个是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,该结构单元的数量为2以下。
[29][21]至[28]任意一项的环状苯亚甲基缩醛连接基团化合物,其中构成X3和X4中的任意一个的官能团包含保护性基团。
本发明的优点
在根据本发明的具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物中,能够根据生命体内弱酸性环境的pH控制环状苯亚甲基缩醛连接基团的水解速率,并且能够在目标部分的pH下可选择地将亲水性聚合物链从连接至亲水性聚合物衍生物的药物等脱附。因此,通过在连接至亲水性聚合物衍生物的生物官能性分子或药物载体被运送至作为目标的组织或位点后脱附亲水性聚合物链,能够从根本上消除例如由形成亲水性聚合物的水合层造成细胞内摄取和核内体逸出的抑制的问题,这些问题是传统的亲水性聚合物改性的缺点。即,通过在药物等的化学改性中使用亲水性聚合物衍生物,能够仅给予亲水性聚合物改性的优点,例如水溶解度和生物利用度的提高以及血液中的循环时间的延长,而不阻碍药物等的原始功能的表达。
另外,由于在亲水性聚合物衍生物中,亲水性聚合物和环状苯亚甲基连接基团的苯环通过在生命体中稳定的键连接,所以在水解时能够避免释放出亲水性聚合物链或连接的药物等以外的低分子量物质,更具体为低分子量芳香醛类,从而能够消除低分子量物质的影响。
附图说明
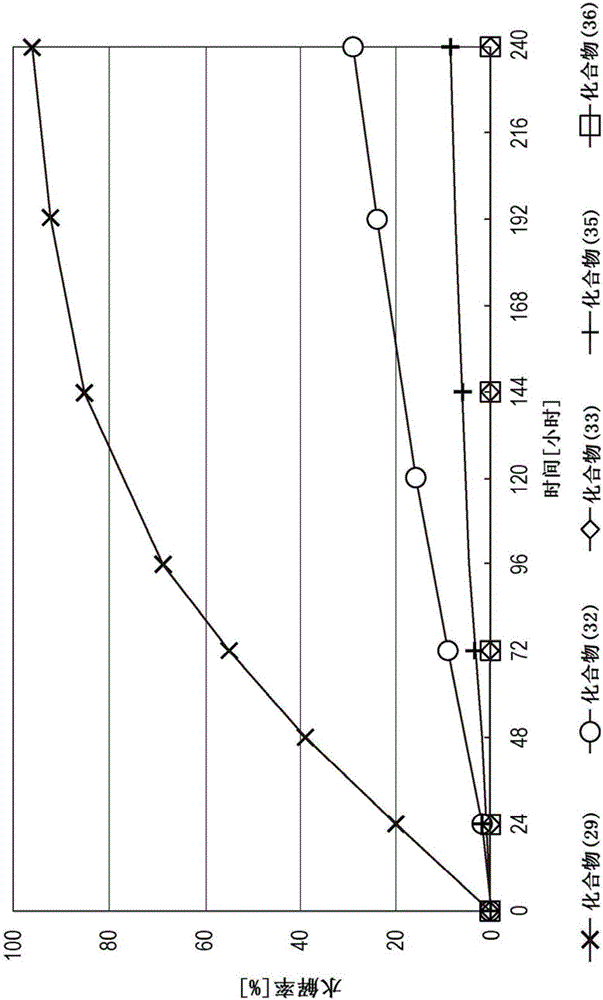
图1示出使用实施例中描述的式(29)、式(32)、式(33)、式(35)和式(36)的化合物,在pD 5.5和37℃下的MES重水缓冲剂中水解测试的结果。
图2示出使用实施例中描述的式(29)、式(32)、式(33)、式(35)和式(36)的化合物,在pD 7.4和37℃下的HEPES重水缓冲剂中水解测试的结果。
具体实施方式
以下,将详细描述本发明。
说明书中使用的术语“缩醛”意指源自醛的缩醛结构和源自酮的缩醛结构两者,源自酮的缩醛结构即缩酮结构。
本文中使用的术语“环状缩醛”意指式(1)中的s是1并且t是0的5元环1,3-二氧戊环结构和式(1)中s是1并且t是1或者s是2并且t是0的6元环1,3-二氧六环结构两者。
本发明的式(1)中的R1和R6各自是氢原子或烃类基团,烃类基团的碳原子数量优选为10以下,并且烃类基团的具体实例包括甲基、乙基、丙基、异丙基、叔丁基、苯基和苯甲基。R1的优选实施方式是氢原子或甲基,并且更优选为氢原子。
本发明的式(1)中的苯环可以具有多个取代基。通过适当地选择苯环上取代基的种类、位置以及给电子性质和吸电子性质的程度,可以调节影响环状缩醛连接基团的水解速率的缩醛基周边的电子密度和立体位阻的程度。这使其可以给予环状缩醛连接基团理想的水解速率。
在本说明书中,使用“取代基常数(σ)”描述式(1)中的苯环上的取代基,取代基常数意指量化取代基对苯衍生物的反应速率或平衡的影响的哈米特法则(Hammett's rule)中的取代基常数。然而,如已知的,哈米特法则仅应用于对位取代和间位取代的苯衍生物,并且不能应用于由立体位阻影响的邻位取代的苯衍生物。因此,在邻位取代的苯衍生物的情况下,取代基常数意指扩展上述哈米特法则的塔夫脱方程(Taft's equation)中的取代基常数。
在上述对位取代和间位取代的苯衍生物中,哈米特法则由以下公式(7)所表示:
log(k/k0)=ρσ (7)
其中k是对位取代和间位取代的苯衍生物的任意反应的速率常数或平衡常数,k0是在苯衍生物不具有任何取代基、即取代基是氢原子的情况下的速率常数或平衡常数,ρ是反应常数,并且σ是取代基常数。
上述式(7)中的反应常数(ρ)是取决于例如反应类型、温度或溶剂的反应条件确定的常数,并且能够从哈米特图(Hammett plot)的斜率计算。在本发明的具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物的酸水解反应中,在1,3-二氧戊环结构的情况下,该常数从对实施例中描述的式(29)、式(32)和式(33)的化合物进行的水解测试的结果中计算为“ρ=-2.7”。而且,在1,3-二氧六环结构的情况下,该常数从对式(35)和式(36)的化合物进行的水解测试的结果中计算为“ρ=-4.8”。
上述式(7)中的取代基常数(σ)是仅取决于取代基的类型和位置确定的常数,与反应类型无关。在不存在取代基、即取代基是氢原子的情况下,该常数是“0”。说明书中使用的术语“吸电子”意指σ是正值的情况,并且术语“给电子”意指σ是负值的情况。
如上所述,哈米特法则仅应用于对位取代或间位取代的苯衍生物,并且不能应用于受立体位阻影响的邻位取代的苯衍生物的情况。因此,塔夫脱方程是将这种立体位阻的影响作为位置因素、即取代基的位置常数(Es)引入,以扩展哈米特法则,使其能够应用于邻位取代的苯衍生物的情况。塔夫脱方程由以下公式(8)所表示。
log(k/k0)=ρ*σ*+Es (8)
其中k是对位取代或间位取代的苯衍生物的任意反应的速率常数或平衡常数,k0是在苯衍生物不具有任何取代基、即取代基是氢原子的情况下的速率常数或平衡常数,ρ*是反应常数,σ*是取代基常数,并且Es是取代基的位置常数。
如已知的,由于对位取代的或间位取代的苯衍生物的反应常数(ρ)和邻位取代苯衍生物的反应常数(ρ*)大体上相等,说明书中定义ρ和ρ*相同。由于邻位的取代基常数(σ*)与对位的取代基常数相似,例如在“Charton,M.Can.J.Chem.1960,38 2493-2499”中所描述的,在说明书中,将对位的相应的取代基常数应用于邻位的取代基常数。
在“Hansch,C.;Leo,A.;Taft,R.W.Chem.Rev.1991,91,165-195”中描述了对位和间位的取代基常数(σ),并且对于取代基常数(σ)未知的取代基,该常数可以通过在“Hammett,L.P.Chem.Rev.1935,17(1),125-136”中描述的方法测量和确定。此外,在“Unger,S.H.;Hansch,C.Prog.Phys.Org.Chem.1976,12,91-118”中描述了位置常数(Es)。然而,对于说明书中使用的Es,氢原子定义为“0”。
在式(1)中,在苯环上存在多个取代基的情况下,定义加成性由其取代基常数(σ)和位置常数(Es)所确立,并且σ的总和由“Σσ”表示并且Es的总和由“ΣEs”所表示。
Z1连接至环状苯亚甲基缩醛的苯环,并且P-Z1也是苯环的取代基。P-Z1的取代基常数能够通过分别测量P的组成和聚合度,并将其与Z1组合来确定,但是因为P-Z1的取代基常数实质上受苯环连接部分周边结构的影响大,其他部分的效果非常小从而可以忽略。因此,可以使用与苯环连接部分周边结构相似的结构的已知取代基常数,来代替分别测量P-Z1的取代基常数。
如本文中所定义,能够使用其中用氢原子取代除连接至从P-Z1主链的骨架原子的苯环连接的原子计算的第三个原子的第二个原子以外的原子的结构的取代基常数,来代替说明书中P-Z1的取代基常数。然而,在用氢原子取代原子而形成羧基的情况下,所定义的是,能够使用其中用甲基代替氢原子来取代原子的结构的取代基常数,来代替P-Z1的取代基常数。
P-Z1的苯环连接部分的结构及用于取代的结构的具体实例在下面示出。在下面示出的(r1)的情况下,其中P-Z1的苯环连接部分是醚键,应用下面示出的(r2)的取代基常数。在下面示出的(r3)和(r5)的情况下,其中P-Z1的苯环连接部分是酰胺键,分别应用下面示出的(r4)和(r6)的取代基常数。在下面示出的(r7)的情况下,其中P-Z1的苯环连接部分是氨基甲酸酯键,应用下面示出的(r8)的取代基常数。
关于本发明的具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物的适当的水解速率,在pH 5.5及37℃的缓冲剂中的水解半衰期(t1/2)优选为在1小时至6个月的范围内,更优选为在1小时至1个月的范围内,并且还更优选为在1小时至24小时的范围内。在本说明书中,使用源自实施例中描述的式(32)的化合物的数值,其中在上述水解条件下的t1/2是12小时,从而限定了包括1,3-二氧戊环结构的情况下的取代基常数的总和(Σσ)的适当的范围。当使用上述方程(7)计算式(32)的化合物的log(k/k0)时,获得了以下等式(9)。然而,根据以上定义,式(32)的化合物中的P-Z1用乙氧基(CH3CH2O-)来代替。
log(k/k0)=-2.7×(0.34-0.24)=-0.27 (9)
在式(1)中的R2和R5是氢原子的情况下,使用以上等式(9)和(7)取t1/2为24小时时的的速率常数作为k'计算log(k'/k0)时,获得了以下等式(10)。
log(k'/k)=log{(12/24)k/k}=-0.30
变形该等式,
log(k'/k)=log[(k'/k0)/(k/k0)]=-0.30
log(k'/k0)-log(k/k0)=-0.30
将以上式(9)代入,
log(k'/k0)-(-0.27)=-0.30
log(k'/k0)=-0.57(10)
此处,当使用以上等式(10)和等式(7)计算取代基常数的总和(Σσ)时,获得了以下等式(11)。
log(k'/k0)=-2.7×Σσ=-0.57
Σσ=0.21 (11)
类似地,在式(1)中的R2和R5是氢原子的情况下,当取t1/2为1小时时的速率常数作为k"计算log(k"/k0)时,获得了以下等式(12)。
log(k"/k)=log(12k/k)=1.08
变形该等式,
log(k"/k)=log[(k"/k0)/(k/k0)]=1.08
log(k"/k0)-log(k/k0)=1.08
将以上式(9)代入,
log(k"/k0)-(0.27)=1.08
log(k"/k0)=0.81 (12)
此处,当使用以上等式(12)和等式(7)计算取代基常数的总和(Σσ)时,获得了以下等式(13)。
log(k"/k0)=-2.7×Σσ=0.81
Σσ=-0.30 (13)
从等式(11)和等式(13),在式(1)包括1,3-二氧戊环结构并且R2和R5是氢原子的情况下,在Σσ的范围满足-0.30≤Σσ≤0.21时,亲水性聚合物衍生物的t1/2为1小时≤t1/2≤24小时。类似地,在1小时≤t1/2≤1个月以及1小时≤t1/2≤6个月时计算的Σσ范围分别得出以下:1小时≤t1/2≤1个月时-0.30≤Σσ≤0.76,1小时≤t1/2≤6个月时-0.30≤Σσ≤1.05。
能够用于本发明中的取代基是不抑制亲水性聚合物衍生物的合成过程中的环状苯亚甲基缩醛连接基团化合物的缩醛化反应、环状苯亚甲基缩醛连接基团化合物与亲水性聚合物中间体的偶联反应、以及亲水性聚合物衍生物的末端官能团转化反应的取代基,并且还不抑制亲水性聚合物衍生物与药物等的成键反应。
取代基可以是任意的吸电子取代基和给电子取代基,只要其满足上述条件即可,并且取代基可以单独使用或组合使用。吸电子取代基包括具有2至5个碳原子的酰基、具有2至5个碳原子的烷氧基羰基、具有2至5个碳原子的氨甲酰基、具有2至5个碳原子的酰氧基、具有2至5个碳原子的酰胺基、具有2至5个碳原子的烷氧基羰基氨基、氟原子、氯原子、溴原子、碘原子、具有1至4个碳原子的烷基巯基、具有1至4个碳原子的烷基磺酰基、具有6至10个碳原子的芳基磺酰基、硝基、三氟甲基、氰基,并且其优选实例包括乙酰基、甲氧基羰基、甲基氨甲酰基、乙酰氧基、乙酰胺基、甲氧基羰基氨基、氟原子、氯原子、溴原子、碘原子、甲基巯基、苯磺酰基、硝基、三氟甲基和氰基。给电子取代基包括具有1至4个碳原子的烷基,并且其优选实例包括甲基、乙基、丙基、异丙基和叔丁基。作为处于间位的吸电子基和处于对位和临位的给电子基团的取代基包括具有1至4个碳原子的烷氧基、具有6至10个碳原子的芳基和具有6至10个碳原子的芳氧基,并且其优选实例包括甲氧基、乙氧基、丙氧基、异丙氧基、叔丁氧基、苯基和苯氧基。
在式(1)包括1,3-二氧戊环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,通过使用在上述取代基中立体位阻的影响最大的苯基和立体位阻的影响最小的氟原子的位置常数(Es),使用塔夫脱方程(8)分别计算在1小时≤t1/2≤24小时、1小时≤t1/2≤1个月以及1小时≤t1/2≤6个月在pH 5.5和37℃的缓冲剂中Σσ的范围,结果分别如下:在1小时≤t1/2≤24小时时,-1.71≤Σσ≤0.04,在1小时≤t1/2≤1个月时,-1.71≤Σσ≤0.59,以及在1小时≤t1/2≤6个月时,-1.71≤Σσ≤0.88。
在式(1)包括1,3-二氧戊环结构并且R2和R5是氢原子的情况下,例如,下面描述了在1小时≤t1/2≤24小时时满足-0.30≤Σσ≤0.21的优选实施方式。然而,此处示出的取代基意指R3和R4以及根据上述定义的代替P-Z1使用的结构。在优选的实施方式中,式(1)中的一个间位是甲氧基、乙氧基或乙酰胺基,并且更优选为乙氧基或乙酰胺基。在另一优选的实施方式中,式(1)中的对位是甲氧基或乙氧基,并且一个间位是独立地选自由氟原子、氯原子、溴原子和碘原子所组成的组中的取代基,并且更优选为对位是甲氧基且一个间位是氟原子或氯原子。在又一优选的实施方式中,式(1)中的对位和间位中的一个是甲氧基、乙氧基或乙酰胺基,并且更优选为甲氧基或乙氧基。
此外,在式(1)包括1,3-二氧戊环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,例如,下面描述了在1小时≤t1/2≤24小时时满足-1.71≤Σσ≤0.04的优选实施方式。然而,此处示出的取代基意指R3和R4以及根据上述定义的代替P-Z1使用的结构。在式(1)中的R2和R5中的一个是氟原子、甲基或乙基并且另一个是氢原子的情况下,对位优选为乙氧基或乙酰胺基,并且更优选为乙氧基。在式(1)中的R2和R5中的一个是甲氧基并且另一个是氢原子的情况下,对位优选为选自由甲氧甲基和乙酰胺基所组成的组中的取代基,并且更优选为乙酰胺基。
此外,使用源自实施例中描述的式(35)的化合物的数值,其中在pH 5.5和37℃的缓冲剂中的水解半衰期(t1/2)是24小时,能够限定式(1)包括1,3-二氧六环结构的情况下的取代基常数的总和(Σσ)的适当的范围。
在式(1)包括1,3-二氧六环结构并且R2和R5是氢原子的情况下,在Σσ的范围满足-0.19≤Σσ≤0.10时,亲水性聚合物衍生物的t1/2为1小时≤t1/2≤24小时。类似地,在1小时≤t1/2≤1个月以及1小时≤t1/2≤6个月时计算的Σσ范围分别得出以下:1小时≤t1/2≤1个月时-0.19≤Σσ≤0.41,1小时≤t1/2≤6个月时-0.19≤Σσ≤0.57。
另外,在式(1)包括1,3-二氧六环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,通过使用在上述取代基中立体位阻的影响最大的苯基和立体位阻的影响最小的氟原子的位置常数(Es),使用塔夫脱方程(8)分别计算在1小时≤t1/2≤24小时、1小时≤t1/2≤1个月以及1小时≤t1/2≤6个月在pH 5.5和37℃的缓冲剂中Σσ的范围。结果分别如下:在1小时≤t1/2≤24小时时,-0.98≤Σσ≤0.00,在1小时≤t1/2≤1个月时,-0.98≤Σσ≤0.31,以及在1小时≤t1/2≤6个月时,-0.98≤Σσ≤0.48。
如上所述,适合于给予本发明的具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物期望的水解能力的取代基的类型和位置能够通过使用等式(7)和等式(8)进行上述计算而合理地设定。
对本发明的式(1)中的X1没有特别限定,只要其是通过与作为化学改性目标的例如生理活性的蛋白质、肽、抗体、核酸或低分子量药物或者例如脂质体和聚合物胶团的药物载体的生物官能性分子中存在的官能团反应形成共价键的官能团即可。例如,官能团包括在“Harris,J.M.Poly(Ethylene Glycol)Chemistry;Plenum Press:New York,1992”、“Hermanson,G.T.Bioconjugate Techniques,2nd ed.;Academic Press:San Diego,CA,2008”和“PEGylated Protein Drugs:Basic Science and Clinical Applications;Veronese,F.M.,Ed.;Birkhauser:Basel,Switzerland,2009”等中描述的官能团。
X1的优选的实例包括活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基和叠氮基。更具体地,能够通过与生物官能性分子的氨基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基或羧基,能够通过与生物官能性分子的巯基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基或乙烯基,能够通过与生物官能性分子的醛基或羧基反应形成共价键的官能团是巯基、氨基、氧氨基或酰肼基,能够通过与生物官能性分子的炔基反应形成共价键的官能团是巯基或叠氮基,并且能够通过与生物官能性分子的叠氮基反应形成共价键的官能团是炔基。
本文中所称的术语“活性酯”指代由式:-C(=O)-L表示的活化羰基,其中L表示离去基团。由L表示的离去基团包括琥珀酰亚胺基氧基,邻苯二甲酰亚胺基氧基、4-硝基苯氧基、1-咪唑基、五氟苯氧基、苯并三唑-1-基氧基和7-氮杂苯并三唑1-基-氧基等。本文中所称的术语“活性碳酸酯”指代由式:-O-C(=O)-L表示的活化碳酸酯基,并且L表示与上述相同的离去基团。
在本发明的优选的实施方式中,X1是由基团(I)、基团(II)、基团(III)、基团(IV)或基团(V)表示的基团。
基团(I):能够通过与生物官能性分子的氨基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)和(f)
基团(II):能够通过与生物官能性分子的巯基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)和(j)
基团(III):能够通过与生物官能性分子的醛基或羧基反应形成共价键的官能团
下列(g)、(k)、(l)和(m)
基团(IV):能够通过与生物官能性分子的炔基反应形成共价键的官能团
下列(g)、(k)、(l)、(m)和(n)
基团(V):能够通过与生物官能性分子的叠氮基反应形成共价键的官能团
下列(j)
在上式中,R7是氢原子或磺基,磺基的具体实例包括磺酸钠和磺酸钾,并且R7优选为氢原子。R8和R11各自是氢原子或具有1至5个碳原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基和戊基。R9是具有1至10个碳原子的可以包含卤素原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、异戊基、己基、苯甲基、4-甲基苯基、三氟甲基、2,2,2-三氟乙基、4-(三氟甲氧基)苯基、乙烯基、氯乙基、溴乙基和碘乙基,并且R9优选为甲基、乙烯基、4-甲基苯基或2,2,2-三氟乙基。R10是选自氯原子、溴原子和碘原子的卤素原子。
本发明的式(1)中的Z1是在环状苯亚甲基缩醛基的苯环和亲水性聚合物链之间的二价间隔物,并且Z2是官能团X1和环状苯亚甲基缩醛基之间的的二价间隔物。它们由共价键组成并且没有特别限定,只要它们对酸水解比环状苯亚甲基缩醛基更稳定即可,并且优选的是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基。亚烷基的碳原子数量优选为从1至24。为了便于说明并且不加以限制,亚烷基的优选实例包括诸如(z1)的结构。具有醚键的亚烷基的优选的实例包括诸如(z2)或(z3)的结构。具有酯键的亚烷基的优选的实例包括诸如(z4)的结构。具有碳酸酯键的亚烷基的优选的实例包括诸如(z5)的结构。具有氨基甲酸乙酯键的亚烷基的优选的实例包括诸如(z6)的结构。具有酰胺键的亚烷基的优选的实例包括诸如(z7)的结构。具有仲氨基的亚烷基的优选的实例包括诸如(z8)的结构。在优选的实施方式中,p和q各自独立地是1至12的整数。例如,在意欲在例如蛋白质内部的疏水性环境中连接官能团X1的情况下,p和q优选为大,并且在意欲在亲水性环境中连接官能团X1的情况下,p和q优选为小。然而,在Z1和Z2的至少一个是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,上述结构单元的数量为2以下。
本发明的式(1)中的P是亲水性聚合物,并且其具体实例包括聚亚烷基二醇、聚噁唑啉、聚碳酸酯、聚氨酯、聚乙烯醇、聚丙烯酸酯、聚甲基丙烯酸酯、聚丙烯酰胺、聚乙烯吡咯烷酮、聚乳酸、聚乙醇酸、聚氨基酸和源自以上聚合物的共聚物,并且P优选为聚亚烷基二醇,更优选为聚乙二醇。
说明书中使用的术语“聚乙二醇”意指具有通过氧化乙烯的聚合获得的分子量分布的聚乙二醇和具有单一分子量的低聚乙二醇通过偶联反应连接获得的单分布聚乙二醇两者。
在本发明的一个方面,式(1)中的P是直线型聚乙二醇。
在此方面的优选的实施方式中,式(1)中的P由式(2)表示。
Y-(OCH2CH2)n- (2)
在该式中,n是每个聚乙二醇链的重复单元的数量,并且在具有分子量分布的聚乙二醇中,定义n是基于化合物的数量平均分子量(Mn)通过各种理论计算而得。
在该式中,Y是具有1至24个碳原子的烃基,其具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、异戊基、己基、庚基、2-乙基己基、辛基、壬基、癸基、十一烷基、十二烷基、十三烷基、十四烷基、十五烷基、十六烷基、十七烷基、十八烷基、十九烷基、二十烷基、二十一烷基、二十二烷基、二十三烷基、二十四烷基、苯基、苯甲基、甲苯基、丁基苯基、十二烷基苯基和三苯甲基,并且Y优选为具有1至10个碳原子的烃基,更优选为甲基或乙基,还更优选为甲基。
在此方面的另一个优选的实施方式中,式(1)中的P由式(3)表示。
X2-Z3-(OCH2CH2)n- (3)
在该式中,X2是不同于X1的化学反应性官能团,并且Z3是在官能团X2和聚乙二醇链之间的二价连接物。由于聚乙二醇衍生物具有两个不同的化学反应性官能团X1和X2,其可以提供具有目标指向性质的聚乙二醇-药物缀合物,例如通过将药物连接至X1并且将目标指向分子连接至X2。
X2的优选的实例包括活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基和叠氮基。更具体地,能够通过与生物官能性分子的氨基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基或羧基,能够通过与生物官能性分子的巯基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基或乙烯基,能够通过与生物官能性分子的醛基或羧基反应形成共价键的官能团是巯基、氨基、氧氨基或酰肼基,能够通过与生物官能性分子的炔基反应形成共价键的官能团是巯基或叠氮基,并且能够通过与生物官能性分子的叠氮基反应形成共价键的官能团是炔基。
在本发明的优选的实施方式中,X2是由基团(I)、基团(II)、基团(III)、基团(IV)或基团(V)表示的基团。
基团(I):能够通过与生物官能性分子的氨基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)和(f)
基团(II):能够通过与生物官能性分子的巯基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)和(j)
基团(III):能够通过与生物官能性分子的醛基或羧基反应形成共价键的官能团
下列(g)、(k)、(l)和(m)
基团(IV):能够通过与生物官能性分子的炔基反应形成共价键的官能团
下列(g)、(k)、(l)、(m)和(n)
基团(V):能够通过与生物官能性分子的叠氮基反应形成共价键的官能团
下列(j)
在上式中,R7是氢原子或磺基,磺基的具体实例包括磺酸钠和磺酸钾,并且R7优选为氢原子。R8和R11各自是氢原子或具有1至5个碳原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基和戊基。R9是具有1至10个碳原子的可以包含卤素原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、异戊基、己基、苯甲基、4-甲基苯基、三氟甲基、2,2,2-三氟乙基、4-(三氟甲氧基)苯基、乙烯基、氯乙基、溴乙基和碘乙基,并且R9优选为甲基、乙烯基、4-甲基苯基或2,2,2-三氟乙基。R10是选自氯原子、溴原子和碘原子的卤素原子。
必要的是,X2不同于X1。作为X1和X2的组合的优选实例,当X1是活性酯基或活性碳酸酯基时,X2是选自马来酰亚胺基、乙烯基砜基、α-卤代乙酰基、炔基和叠氮基的基团;当X1是醛基时,X2是选自马来酰亚胺基、乙烯基砜基、炔基和叠氮基的基团;当X1是马来酰亚胺基、乙烯基砜基或α-卤代乙酰基时,X2是选自活性酯基、活性碳酸酯基、炔基和叠氮基的基团;当X1是炔基或叠氮基时,X2是选自马来酰亚胺基、乙烯基砜基、α-卤代乙酰基、活性酯基、活性碳酸酯基、氨基和氧氨基的基团;当X1是氨基或氧氨基时,X2是炔基、叠氮基、巯基或羧基;并且当X1是巯基时,X2是选自氨基、氧氨基、叠氮基和羧基的基团。更优选地,当X1是活性酯基或活性碳酸酯基时,X2是选自马来酰亚胺基、α-卤代乙酰基、炔基和叠氮基的基团;当X1是醛基时,X2是选自马来酰亚胺基、α-卤代乙酰基、炔基和叠氮基的基团;当X1是马来酰亚胺基或α-卤代乙酰基时,X2是选自活性酯基、活性碳酸酯基、炔基和叠氮基的基团;当X1是炔基或叠氮基时,X2是选自马来酰亚胺基、α-卤代乙酰基、活性酯基、活性碳酸酯基、氨基和氧氨基的基团;当X1是氨基或氧氨基时,X2是炔基、叠氮基或巯基;并且当X1是巯基时,X2是选自氨基、氧氨基和叠氮基的基团。
Z3由共价键组成并且没有特别限定,只要其对酸水解比环状苯亚甲基缩醛基更稳定即可,并且优选的是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基。亚烷基的碳原子数量优选为从1至24。为了便于说明并且不加以限制,亚烷基的优选实例包括诸如(z1)的结构。具有醚键的亚烷基的优选的实例包括诸如(z2)或(z3)的结构。具有酯键的亚烷基的优选的实例包括诸如(z4)的结构。具有碳酸酯键的亚烷基的优选的实例包括诸如(z5)的结构。具有氨基甲酸乙酯键的亚烷基的优选的实例包括诸如(z6)的结构。具有酰胺键的亚烷基的优选的实例包括诸如(z7)的结构。具有仲氨基的亚烷基的优选的实例包括诸如(z8)的结构。在优选的实施方式中,p和q各自独立地是1至12的整数。例如,在意欲在例如蛋白质内部的疏水性环境中连接官能团X2的情况下,p和q优选为大,并且在意欲在亲水性环境中连接官能团X2的情况下,p和q优选为小。然而,在Z3是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,上述结构单元的数量为2以下。
在本发明的另一个方面,式(1)中的P是分枝型聚乙二醇。
在此方面的优选的实施方式中,式(1)中的P由式(4)表示。
在该式中,Y是如上所述的具有1至24个碳原子的烃基,并且v是0或2。
在v是0的情况下存在两条聚乙二醇链,并且在v是2的情况下存在四条聚乙二醇链。一般来说,在用聚乙二醇对生物相关物质的化学改性中,当引入多于需要的聚乙二醇的连接点时,破坏了生物相关物质的活性位点从而减少其功能,因此进行了通过增加聚乙二醇的分子量来提高效果的尝试。然而,粘度随着分子量的增加而增加,并且因此例如,作为诸如注射剂型的水溶液剂型的操作变得困难。由于聚乙二醇衍生物具有分枝型结构,与具有相同分子量的直线型聚乙二醇衍生物相比,其显示出低粘度,因此在例如水溶液剂型的应用中是有用的。
在此方面的另一个优选的实施方式中,式(1)中的P由式(5)表示。
在该式中,X2是如上所述的不同于X1的化学反应性官能团,Z3是如上所述的二价间隔物,并且v是0或2。
聚乙二醇衍生物具有一个X1和两个或四个X2,并且例如当药物连接至X1并且目标指向分子连接至X2时,能够获得高的目标指向性能。
在此方面的又一个优选的实施方式中,式(1)中的P由式(6)表示。
在该式中,X2是如上所述的不同于X1的化学反应性官能团,Z3是如上所述的二价间隔物,并且v是0或2。
在抗体-药物缀合物(ADC)相关领域中,为了提高药物运送效率,优选的是将多个药物连接至抗体,但是当向抗体中引入多个连接点时,与抗原的亲和性的下降成为问题。聚乙二醇衍生物具有两个或四个X1和一个X2,并且例如在靶向癌症的ADC中,当抗癌药剂连接至X1并且抗体连接至X2时,可以在不增加抗体的连接点的同时改善抗癌药剂的运送效率。
在本发明的又一个方面,式(1)中的P是具有2至8的末端数的聚乙二醇,构成P的聚乙二醇全部末端各自结合于Z1,并且w等于聚乙二醇的末端数。
在此方面的优选的实施方式中,式(1)中的P选自由式(r)、式(s)、式(t)、式(u)和式(v)所组成的组。在P由式(r)表示的情况下w是2,在P由式(s)表示的情况下w是3,在P由式(t)表示的情况下w是4,在P由式(u)表示的情况下w是4,并且在P由式(v)表示的情况下w是8。
本发明的式(2)和(3)中的n的优选范围是从3至2000,更优选为从20至1500,还更优选为从40至1000,并且最优选为从60至500。另外,式(4)、式(5)和式(6)中的n的优选范围是从3至1000,优选为从10至800,更优选为从20至500,并且最优选为从30至300。
根据本发明的另一个方面,提供了一种由式(55)表示的环状苯亚甲基缩醛连接基团化合物。
本发明的式(55)中的R1和R6各自是氢原子或烃类基团,烃类基团的碳原子数量优选为10以下并且烃类基团的具体实例包括甲基、乙基、丙基、异丙基、叔丁基、苯基和苯甲基。R1的优选实施方式是氢原子或甲基,并且更优选为氢原子。
本发明的式(55)中的苯环可以具有多个取代基。通过适当地选择苯环上取代基的种类、位置以及给电子性质和吸电子性质的程度,可以调整影响环状缩醛连接基团的水解速率的缩醛基周边的电子密度和立体位阻的程度。这使其可以给予环状缩醛连接基团理想的水解速率。
在本说明书中,使用“取代基常数(σ)”描述式(55)中的苯环上的取代基,取代基常数意指量化取代基对苯衍生物的反应速率或平衡的影响的哈米特法则(Hammett's rule)中的取代基常数。然而,如已知的,哈米特法则仅应用于对位取代和间位取代的苯衍生物,并且不能应用于受立体位阻影响的邻位取代的苯衍生物。因此,在邻位取代的苯衍生物的情况下,取代基常数意指扩展上述哈米特法则的塔夫脱方程(Taft's equation)中的取代基常数。
如已知的,由于哈米特法则中对位取代的或间位取代的苯衍生物的反应常数(ρ)和塔夫脱方程中邻位取代苯衍生物的反应常数(ρ*)大体上相等,说明书中定义ρ和ρ*相同。由于塔夫脱方程中邻位的取代基常数(σ*)与对位的取代基常数相似,例如在“Charton,M.Can.J.Chem.1960,38 2493-2499”中所描述,在说明书中,将对位的相应的取代基常数应用于邻位的取代基常数。
在“Hansch,C.;Leo,A.;Taft,R.W.Chem.Rev.1991,91,165-195”中描述了对位和间位的取代基常数(σ),并且对于取代基常数(σ)未知的取代基,该常数可以通过在“Hammett,L.P.Chem.Rev.1935,17(1),125-136”中描述的方法测量和确定。此外,在“Unger,S.H.;Hansch,C.Prog.Phys.Org.Chem.1976,12,91-118”中描述了塔夫脱方程中的位置常数(Es)。然而,本文中使用的Es是氢原子定义为“0”的Es。
在式(55)中,在苯环上存在多个取代基的情况下,定义加成性由其取代基常数(σ)和位置常数(Es)所确立,并且σ的总和由“Σσ”表示以及Es的总和由“ΣEs”所表示。
Z1连接至环状苯亚甲基缩醛的苯环,并且X3-Z1也是苯环的取代基。X3-Z1的取代基常数能够通过分别测量X3和Z1的组合来确定,但是因为X3-Z1的取代基常数实质上受苯环连接部分周边结构的影响大,其他部分的效果非常小从而可以忽略。因此,可以使用与苯环连接部分周边结构相似的结构的已知取代基常数,来代替单独分别测量X3-Z1的取代基常数。
如说明书中所定义,能够使用其中用氢原子取代连接至从X3-Z1主链的骨架原子的苯环连接的原子计算的第三个原子的第二个原子以外的原子的结构的取代基常数,来代替X3-Z1的取代基常数。然而,在用氢原子取代原子而形成羧基的情况下,所定义的是,能够使用其中用甲基代替氢原子取代原子的结构的取代基常数,来代替X3-Z1的取代基常数。此外,在X3-Z1的主链的骨架原子为4个原子以下的情况下,使用上述文献中记载的已知取代基常数,或者能够使用通过根据上述文献中记载的方法测量而获得的值。
X3-Z1的苯环连接部分的结构及用于取代的结构的具体实例在下面示出。在下面示出的(r1)的情况下,其中X3-Z1的苯环连接部分是醚键,应用下面示出的(r2)的取代基常数。在下面示出的(r3)和(r5)的情况下,其中X3-Z1的苯环连接部分是酰胺键,分别应用下面示出的(r4)和(r6)的取代基常数。在下面示出的(r7)的情况下,其中X3-Z1的苯环连接部分是氨基甲酸酯键,应用下面示出的(r8)的取代基常数。
能够用于此方面的取代基是在环状苯亚甲基缩醛连接基团化合物的合成过程中不抑制缩醛化反应和末端官能团转化反应的取代基,并且在本发明的优选实施方式中是不抑制除上述反应以外的连接基团化合物与亲水性聚合物中间体的偶联反应、所获得的亲水性聚合物衍生物的末端官能团转化反应以及亲水性聚合物衍生物和药物等的成键反应的取代基。
取代基可以是任意的吸电子取代基和给电子取代基,只要其满足上述条件即可,并且取代基可以单独使用或组合使用。吸电子取代基包括具有2至5个碳原子的酰基、具有2至5个碳原子的烷氧基羰基基体、具有2至5个碳原子的氨甲酰基、具有2至5个碳原子的酰氧基、具有2至5个碳原子的酰胺基、具有2至5个碳原子的烷氧基羰基氨基、氟原子、氯原子、溴原子、碘原子、具有1至4个碳原子的烷基巯基、具有1至4个碳原子的烷基磺酰基、具有6至10个碳原子的芳基磺酰基、硝基、三氟甲基、氰基,并且其优选实例包括乙酰基、甲氧基羰基、甲基氨甲酰基、乙酰氧基、乙酰胺基、甲氧基羰基氨基、氟原子、氯原子、溴原子、碘原子、甲基巯基、苯磺酰基、硝基、三氟甲基和氰基。给电子取代基包括具有1至4个碳原子的烷基,并且其优选实例包括甲基、乙基、丙基、异丙基和叔丁基。作为处于间位的吸电子基和处于对位和临位的给电子基团的取代基包括具有1至4个碳原子的烷氧基、具有6至10个碳原子的芳基和具有6至10个碳原子的芳氧基,并且其优选实例包括甲氧基、乙氧基、丙氧基、异丙氧基、叔丁氧基、苯基和苯氧基。
关于本发明的环状苯亚甲基缩醛连接基团化合物的水解速率,在pH 5.5及37℃的缓冲剂中的水解半衰期(t1/2)优选为在1小时至6个月的范围内,更优选为在1小时至1个月的范围内,并且还更优选为在1小时至24小时的范围内。
在式(55)包括1,3-二氧戊环结构并且R2和R5是氢原子的情况下,优选的取代基常数总和(Σσ)的范围在1小时≤t1/2≤24小时时满足-0.30≤Σσ≤0.21,在1小时≤t1/2≤1个月时满足-0.30≤Σσ≤0.76,以及在1小时≤t1/2≤6个月时满足-0.30≤Σσ≤1.05。
在式(55)包括1,3-二氧戊环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,Σσ的范围在1小时≤t1/2≤24小时时满足-1.71≤Σσ≤0.04,在1小时≤t1/2≤1个月时满足-1.71≤Σσ≤0.59,以及在1小时≤t1/2≤6个月时满足-1.71≤Σσ≤0.88。
在式(55)包括1,3-二氧戊环结构并且R2和R5是氢原子的情况下,例如,下面描述了在1小时≤t1/2≤24小时时满足-0.30≤Σσ≤0.21的优选实施方式。然而,此处示出的取代基意指R3、R4和X3-Z1以及根据上述定义的代替X3--Z1使用的结构。在优选的实施方式中,式(55)中的一个间位是甲氧基、乙氧基或乙酰胺基,并且更优选为乙氧基或乙酰胺基。在另一优选的实施方式中,式(55)中的对位是甲氧基或乙氧基,并且一个间位是选自由氟原子、氯原子、溴原子和碘原子所组成的组中的取代基,并且更优选为对位是甲氧基且一个间位是氟原子或氯原子。在又一优选的实施方式中,式(55)中的对位和间位中的一个是甲氧基、乙氧基或乙酰胺基,并且更优选为甲氧基或乙氧基。
此外,在式(55)包括1,3-二氧戊环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,例如,下面描述了在1小时≤t1/2≤24小时时满足-1.71≤Σσ≤0.04的优选实施方式。然而,此处示出的取代基意指R3、R4和X3-Z1以及根据上述定义的代替X3-Z1使用的结构。在式(55)中的R2和R5中的一个是氟原子、甲基或乙基并且另一个是氢原子的情况下,对位优选为乙氧基或乙酰胺基,并且更优选为乙氧基。在式(55)中的R2和R5中的一个是甲氧基并且另一个是氢原子的情况下,对位优选为选自由甲氧甲基和乙酰胺基所组成的组中的取代基,并且更优选为乙酰胺基。
在式(55)包括1,3-二氧六环结构并且R2和R5是氢原子的情况下,优选的取代基常数总和(Σσ)的范围在1小时≤t1/2≤24小时时满足-0.19≤Σσ≤0.10,在1小时≤t1/2≤1个月时满足-0.19≤Σσ≤0.41,以及在1小时≤t1/2≤6个月时满足-0.19≤Σσ≤0.57。
另外,在式(55)包括1,3-二氧六环结构并且R2和R5的至少一个是氢原子以外的取代基的情况下,Σσ的范围在1小时≤t1/2≤24小时时满足-0.98≤Σσ≤0.00,在1小时≤t1/2≤1个月时满足-0.98≤Σσ≤0.31,以及在1小时≤t1/2≤6个月时满足-0.98≤Σσ≤0.48。
式(55)中的X3和X4各自独立地是化学反应性官能团,并且尽管无意限定连接基团化合物的应用,在本发明的优选实施方式中,具有式(1)的环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物能够通过X3和亲水性聚合物中间体的化学反应性官能团的之间的偶联反应来合成。
式(55)中的X3和X4的优选的实例包括活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基、乙烯基、氨基、氧氨基、酰肼基、叠氮基和羟基。根据更具体的实施方式,能够通过与配合反应物(reaction partner)的氨基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基或羧基;能够通过与与配合反应物的巯基反应形成共价键的官能团是活性酯基、活性碳酸酯基、醛基、异氰酸酯基、异硫氰酸酯基、环氧基、马来酰亚胺基、乙烯基砜基、丙烯酸基、磺酰氧基、羧基、巯基、二硫代吡啶基、α-卤代乙酰基、炔基、烯丙基或乙烯基;能够通过与与配合反应物的醛基或羧基反应形成共价键的官能团是巯基、氨基、氧氨基或酰肼基;能够通过与配合反应物的炔基反应形成共价键的官能团是巯基或叠氮基,能够通过与配合反应物的叠氮基反应形成共价键的官能团是炔基;并且能够通过与配合反应物的卤代烷基、烷基磺酸酯或芳基磺酸酯反应形成共价键的官能团是羟基、巯基或氨基。
在此方面的优选实施方式中,X3和X4各自是由基团(I)、基团(II)、基团(III)、基团(IV)、基团(V)或基团(VI)表示的基团。
基团(I):能够通过与配合反应物的氨基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)和(f)
基团(II):能够通过与配合反应物的巯基反应形成共价键的官能团
下列(a)、(b)、(c)、(d)、(e)、(f)、(g)、(h)、(i)和(j)
基团(III):能够通过与配合反应物的醛基或羧基反应形成共价键的官能团
下列(g)、(k)、(l)和(m)
基团(IV):能够通过与配合反应物的炔基反应形成共价键的官能团
下列(g)、(k)、(l)、(m)和(n)
基团(V):能够通过与配合反应物的叠氮基反应形成共价键的官能团
下列(j)
基团(VI):能够通过与配合反应物的卤代烷基、烷基磺酸酯或芳基磺酸酯反应形成共价键的官能团
下列(o)、(g)和(k)
在上式中,R7是氢原子或磺基,磺基的具体实例包括磺酸钠和磺酸钾,并且R7优选为氢原子。R8和R11各自是氢原子或具有1至5个碳原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基和戊基。R9是具有1至10个碳原子的可以包含卤素原子的烃基,并且烃基的具体实例包括甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、异戊基、己基、苯甲基、4-甲基苯基、三氟甲基、2,2,2-三氟乙基、4-(三氟甲氧基)苯基、乙烯基、氯乙基、溴乙基和碘乙基,并且R9优选为甲基、乙烯基、4-甲基苯基或2,2,2-三氟乙基。R10是选自氯原子、溴原子和碘原子的卤素原子。
X3和X4可以彼此相同或不同。作为互不相同的X3和X4的组合的优选实例,当X3是活性酯基或活性碳酸酯基时,X4是选自马来酰亚胺基、乙烯基砜基、α-卤代乙酰基、炔基和叠氮基的基团;当X3是醛基时,X4是选自马来酰亚胺基、乙烯基砜基、炔基和叠氮基的基团;当X3是马来酰亚胺基、乙烯基砜基或α-卤代乙酰基时,X4是选自活性酯基、活性碳酸酯基、炔基和叠氮基的基团;当X3是炔基或叠氮基时,X4是选自马来酰亚胺基、乙烯基砜基、α-卤代乙酰基、活性酯基、活性碳酸酯基、氨基、氧氨基和羟基的基团;当X3是氨基或氧氨基时,X4是炔基、叠氮基、巯基、羟基或羧基;并且当X3是巯基或羟基时,X4是选自氨基、氧氨基、叠氮基和羧基的基团。更优选地,当X3是活性酯基或活性碳酸酯基时,X4是选自马来酰亚胺基、α-卤代乙酰基、炔基和叠氮基的基团;当X3是醛基时,X4是选自马来酰亚胺基、α-卤代乙酰基、炔基和叠氮基的基团;当X3是马来酰亚胺基或α-卤代乙酰基时,X4是选自活性酯基、活性碳酸酯基、炔基和叠氮基的基团;当X3是炔基或叠氮基时,X4是选自马来酰亚胺基、α-卤代乙酰基、活性酯基、活性碳酸酯基、氨基、氧氨基和羟基的基团;当X3是氨基或氧氨基时,X4是炔基、叠氮基、羟基或巯基;并且当X3是巯基或羟基时,X4是选自氨基、氧氨基和叠氮基的基团。
此方面的式(55)中的Z1是在环状苯亚甲基缩醛基的苯环和官能团X3之间的二价间隔物,并且Z2是官能团X4和环状苯亚甲基缩醛基之间的的二价间隔物。它们由共价键组成并且没有特别限定,只要它们对酸水解比环状苯亚甲基缩醛基更稳定即可,并且优选的是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基。亚烷基的碳原子数量优选为从1至24。为了便于说明并且不加以限制,亚烷基的优选实例包括诸如(z1)的结构。具有醚键的亚烷基的优选的实例包括诸如(z2)或(z3)的结构。具有酯键的亚烷基的优选的实例包括诸如(z4)的结构。具有碳酸酯键的亚烷基的优选的实例包括诸如(z5)的结构。具有氨基甲酸乙酯键的亚烷基的优选的实例包括诸如(z6)的结构。具有酰胺键的亚烷基的优选的实例包括诸如(z7)的结构。具有仲氨基的亚烷基的优选的实例包括诸如(z8)的结构。在优选的实施方式中,p和q各自独立地是1至12的整数。然而,在Z1和Z2的至少一个是醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基或包含任意的这些键和基团的亚烷基并且连接了多个重复结构单元的情况下,上述结构单元的数量为2以下。
本发明的具有环状苯亚甲基缩醛连接基团的亲水性聚合物衍生物能够通过在具有取代基的环状苯亚甲基缩醛连接基团化合物和亲水性聚合物中间体之间进行偶联反应而合成。通过偶联反应生成的键由反应中使用的官能团的组合而确定,并且是上述二价间隔物Z1中包含的醚键、酯键、碳酸酯键、氨基甲酸乙酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基。在合成的亲水性聚合物衍生物中,末端官能团按照需要化学转化。关于用于官能团转化的反应,可以使用传统已知的方法,但是需要恰当地选择式(1)的环状苯亚甲基缩醛基和上述二价间隔物Z1和Z2中包含的键不被分解的条件。
关于在环状苯亚甲基缩醛连接基团化合物和亲水性聚合物中间体之间进行偶联反应以及末端官能团的化学转化的典型实例,例示了下列步骤。此处描述作为典型亲水性聚合物的聚乙二醇,作为实例。
(A)环状苯亚甲基缩醛连接基团化合物的合成
其中R1是氢原子或烃类基团;并且R2、R3、R4和R5各自独立地是吸电子或给电子取代基或氢原子。
使具有作为化学反应性官能团的羟基的式(14)的羰基化合物与具有苯邻二甲酰亚胺基的氨基被邻苯二甲酰基保护的式(15)的1,2-二醇衍生物在例如甲苯、苯、二甲苯、乙腈、乙酸乙酯、二乙基醚、叔丁基甲基醚、四氢呋喃、氯仿、二氯甲烷、二甲亚砜、二甲基甲酰胺或二甲基乙酰胺的非质子溶剂中或不使用溶剂,在酸催化剂的存在下反应以获得以下式(16)的具有环状苯亚甲基缩醛基团的化合物。所得化合物可以通过萃取、重结晶、吸附剂处理或柱色谱等纯化。替代羰基化合物,可以使用低级醇的对应缩醛衍生物。该低级醇优选为具有1至5个碳原子的醇,并且更优选为甲醇或乙醇。酸催化剂可以是有机酸或无机酸并且没有特别限定,并且其具体实例包括对甲苯磺酸、吡啶对甲苯磺酸盐、甲磺酸、10-樟脑磺酸、盐酸、碘、氯化铵、草酸和三氟化硼-二甲醚络合物等。
本文中所称的“保护基团”是在某些反应条件下阻止或阻碍分子中特定的化学反应性官能团的反应的成分。保护基团取决于要保护的化学反应性官能团的类型、使用的条件和分子中其他官能团或保护基团的存在而变化。保护基团的特定实例能够在许多普通书籍中找到,并且例如在“Wuts,P.G.M.;Greene,T.W.Protective Groups in Organic Synthesis,4th ed.;Wiley-Interscience:New York,2007”中描述的。此外,通过使用适合于各种保护基团的反应条件,即通过发生化学反应,能够使被保护基团保护的官能团通过去保护而再生原始的官能团。因此,在说明书中,被保护基团保护并且能够通过各种反应去保护的官能团包括在“化学反应性官能团”中。保护基团的典型去保护条件记载在上述文献中。
关于式(14)的化合物中的化学反应性官能团,也能够使用羟基以外的官能团。其具体实例包括羟烷基、氨基、氨烷基、羧基和羧烷基。而且,上述官能团可以被保护基团保护,该保护基团在缩醛化反应的酸性条件下稳定,并且能够在分解环状苯亚甲基缩醛连接基团的催化还原以外的反应条件下去保护。作为要被保护的官能团和保护基团的优选组合,当要被保护的官能团是羟基或羟烷基时,例如,例示了甲硅烷基系保护基团和酰基系保护基团,并且其具体实例包括叔丁基二苯基甲硅烷基、叔丁基二甲基甲硅烷基、三异丙基甲硅烷基、乙酰基和三甲基乙酰基。当要被保护的官能团是氨基或氨烷基时,例如,例示了酰基系保护基团和氨基甲酸酯系保护基团,并且其具体实例包括三氟乙酰基、9-芴甲氧羰基和2-(三甲基甲硅烷基)乙氧羰基。当要被保护的官能团是羧基或羧烷基时,例如,例示了烷基酯系保护基团和甲硅烷基酯系保护基团,并且其具体实例包括甲基、9-芴甲基和叔丁基二甲基甲硅烷基。具体保护基团的类型和典型去保护条件记载在上述文献中,并且可以选择适合于各个保护基团的反应条件,并且去保护能够在与亲水性聚合物中间体的反应之前进行。
此外,关于式(15)的化合物中1,2-二醇组成部分之外的化学反应性官能团,也能够使用苯邻二甲酰亚胺基之外的官能团。在化学反应性官能团是被保护基团保护的官能团的情况下,必要的是,该保护基团在缩醛化反应的酸性条件下稳定,并且能够在分解苯亚甲基缩醛连接基团的催化还原以外的反应条件下去保护。作为要被保护的官能团和保护基团的优选组合,当要被保护的官能团是氨基时,例如,例示了酰基系保护基团和氨基甲酸酯系保护基团,并且其具体实例包括三氟乙酰基、9-芴甲氧羰基和2-(三甲基甲硅烷基)乙氧羰基。当要被保护的官能团是羟基时,例如,例示了甲硅烷基系保护基团和酰基系保护基团,并且其具体实例包括叔丁基二苯基甲硅烷基、叔丁基二甲基甲硅烷基、三异丙基甲硅烷基、乙酰基和三甲基乙酰基。当要被保护的官能团是羧基时,例如,例示了烷基酯系保护基团和甲硅烷基酯系保护基团,并且其具体实例包括甲基、9-芴甲基和叔丁基二甲基甲硅烷基。当要被保护的官能团是磺酰基时,例如,例示了硫醚系保护基团、硫代碳酸酯系保护基团和二硫化物系保护基团,并且其具体实例包括S-2,4-二硝基苯基、S-9-芴甲氧羰基和S-叔丁基二硫基。保护基团的典型去保护条件记载在上述文献中,并且可以选择适合于各个保护基团的反应条件。然而,在化学反应性基团是即使不被保护基团保护时也不抑制缩醛化反应的官能团的情况下,不需要使用保护基团。
(B)聚乙二醇中间体的合成
以作为引发剂的甲醇的3至2000摩尔当量的量,在甲苯中或不使用溶剂,在例如金属钠、金属钾、氢化钠或氢化钾的碱性条件下聚合氧化乙烯,以获得式(17)的聚乙二醇。引发剂优选为具有1至24个碳原子的烃基的醇,并且具体包括甲醇、乙醇、丙醇、异丙醇、丁醇、叔丁醇、苯酚和苯甲醇。由于聚乙二醇具有作为化学反应性基团的羟基,其能够在与环状苯亚甲基缩醛连接基团化合物的偶联反应中原样使用。
CH3-(OCH2CH2)n-OH (17)
使式(17)的聚乙二醇与甲磺酰氯在例如甲苯、苯、二甲苯、乙腈、乙酸乙酯、二乙基醚、叔丁基甲基醚、四氢呋喃、氯仿、二氯甲烷、二甲亚砜、二甲基甲酰胺或二甲基乙酰胺的非质子溶剂中或不使用溶剂,在例如三乙胺、N-甲基吗啉、吡啶或4-二甲基氨基吡啶的有机碱或例如碳酸钠、碳酸氢钠、乙酸钠或碳酸钾的无机碱的存在下反应,以获得式(18)的聚乙二醇中间体。可以不使用有机碱和无机碱。对有机碱或无机碱的使用比率没有特别限定,并且优选为式(17)的聚乙二醇的羟基的等摩尔量以上。而且,可以使用有机碱作为溶剂。获得的化合物可以通过例如萃取、重结晶、吸附剂处理、再沉淀、柱色谱或超临界萃取的纯化方法纯化。
关于式(18)的聚乙二醇中间体中的化学反应性官能团,也能够使用其他官能团。化学反应性官能团的优选实例是其中通过聚乙二醇中间体与上述环状苯亚甲基缩醛连接基团化合物的偶联反应生成的键成为式(1)的二价间隔物Z1中包含的醚键、酯键、碳酸酯键、氨基甲酸酯键、酰胺键、仲氨基、包含任意的这些键和基团的亚烷基、单键或亚烷基的官能团,并且具体包括例如卤素原子、活性酯、活性碳酸酯、醛基、氨基、羟基和羧基。
(C)环状苯亚甲基缩醛连接基团化合物与聚乙二醇中间体的偶联反应
将式(16)的苯亚甲基缩醛连接基团化合物与式(18)的聚乙二醇中间体在例如甲苯、苯、二甲苯、乙腈、乙酸乙酯、二乙基醚、叔丁基甲基醚、四氢呋喃、氯仿、二氯甲烷、二甲亚砜、二甲基甲酰胺或二甲基乙酰胺的非质子溶剂中或不使用溶剂,在例如三乙胺、N-甲基吗啉、叔丁醇钾或吡啶或六甲基二硅胺基钠的有机碱或例如碳酸钾、氢氧化钾或氢化钠的无机碱的存在下进行偶联反应,以获得式(19)的化合物。对有机碱或无机碱的使用比率没有特别限定,但是优选为式(18)的聚乙二醇中间体的化学反应性官能团的等摩尔量以上。而且,可以使用有机碱作为溶剂。获得的化合物可以通过前述纯化方法纯化。
可以在与聚乙二醇中间体的偶联反应之前,对环状苯亚甲基缩醛连接基团化合物的化学反应性官能团进行官能团转化。偶联反应的反应条件取决于环状苯亚甲基缩醛连接基团化合物的化学反应性官能团与聚乙二醇中间体的化学反应性官能团的组合,并且能够使用传统已知的方法。然而,需要恰当地选择式(1)的环状苯亚甲基缩醛基团与上述二价间隔物Z1和Z2包含的键不分解的条件。
(D)具有环状苯亚甲基缩醛连接基团的聚乙二醇衍生物的末端官能团的转化
使用例如乙二胺、甲基肼或甲胺的碱性有机化合物或者例如肼、羟胺或氢氧化钠的碱性无机化合物,在例如水、甲醇或乙醇的质子溶剂中,或者在例如乙腈、四氢呋喃、二甲亚砜、二甲基甲酰胺或二甲基乙酰胺的非质子溶剂中或不使用溶剂,处理式(19)的化合物,以获得其中苯邻二甲酰亚胺基去保护并且转化为氨基的式(20)的化合物。对碱性化合物的使用比率没有特别限定,并且优选为式(19)的化合物的化学反应性官能团的等摩尔量以上。而且,可以使用碱性化合物作为溶剂。获得的化合物可以通过前述纯化方法纯化。
另外,使式(20)的化合物与N-琥珀酰亚胺基-3-马来酰亚胺基丙酸酯在例如甲苯、苯、二甲苯、乙腈、乙酸乙酯、二乙基醚、叔丁基甲基醚、四氢呋喃、氯仿、二氯甲烷、二甲亚砜、二甲基甲酰胺或二甲基乙酰胺的非质子溶剂中或不使用溶剂,在例如三乙胺、N-甲基吗啉、吡啶或4-二甲基氨基吡啶的有机碱或例如碳酸钠、碳酸氢钠、乙酸钠或碳酸钾的无机碱的存在下反应,以获得其中向末端引入了马来酰亚胺基的式(21)的化合物。可以不使用有机碱和无机碱。对有机碱或无机碱的使用比率没有特别限定,并且优选为式(20)的化合物的化学反应性官能团的等摩尔量以上。而且,可以使用有机碱作为溶剂。获得的化合物可以通过前述纯化方法纯化。
实施例
将通过参考实施例,进一步具体地描述本发明,但是本发明不被解释为局限于这些实施例。
在1H-NMR分析中,使用了由JEOL DATUM Ltd.制造的JNM-ECP400或JNM-ECA600。在测量中,使用了5mmφ的试管,并且在氘代溶剂为CDCl3、CD3CN或CD3OD的情况下使用四甲基硅烷(TMS)作为内标物,或者在氘代溶剂为D2O的情况下使用HDO作为标准物。
在凝胶渗透色谱(GPC)分析中,使用SHODEX GPC SYSTEM-11作为GPC系统,SHODEX RIX8作为差示析射计即检测器,以及三根色谱柱即SHODEX KF801L、KF803L和KF804L(φ8mm×300mm)串联连接作为GPC色谱柱,并且柱温箱的温度设定为40℃。使用四氢呋喃作为洗脱剂进行测量,流速为1mL/min,样品浓度为0.1wt%,并且进样量为0.1mL。使用由Kanto Chemical Co.,Ltd.制造的乙二醇、二甘醇和三甘醇以及由Polymer Laboratory制造的具有600至70000的分子量的聚乙二醇或聚氧化乙烯的GPC聚合物标准样品,准备校准曲线。关于数据分析,使用了BORWIN GPC计算程序。Mn表示数量平均分子量,Mw表示重量平均分子量,并且分子量分布示出为Mw/Mn的计算值。
基于在“Glasoe,P.K.;Long,F.A.J.Phys.Chem.1960,64,188-190”中描述的下列关系等式,通过向0.1M的MES(2-吗啉乙磺酸)氘代水溶液和0.1M的HEPES(2-[4-(羟乙基)-1-哌嗪基]乙磺酸)氘代水溶液中分别加入0.1M的氢氧化钠氘代水溶液,分别制备了用于水解测试的pD 5.5的MES氘代水缓冲剂和pD 7.4的HEPES氘代水缓冲剂。
pD=pH计的测量值+0.40
由1H-NMR评估水解率,取缩醛基的氢的积分值和通过水解形成的醛基的氢的积分值,分别作为I1和I2,根据下列计算公式进行计算。
水解率(%)=[I2/(I1+I2)]×100
(实施例1)
向配备有温度计、氮气进气管、搅拌器、Dean-stark管和冷凝管的200mL三颈烧瓶中注入1,2,6-己三醇(30.0g,0.224mol)、丙酮缩二甲醇(25.6g,0.246mol)和对甲苯磺酸一水合物(0.426g,2.24mmol),在80℃进行反应3小时同时蒸馏去除甲醇。向其中加入三乙胺(0.453g,4.48mmol)并将混合物搅拌一段时间,用乙酸乙酯稀释,并且用20重量%的氯化钠溶液清洗。有机层用无水硫酸钠干燥,过滤后,在减压下蒸馏去除溶剂。残留物通过硅胶色谱法纯化,以获得式(22)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.35(3H,s,-CH3),1.41(3H,s,-CH3),1.49-1.67(6H,m,>CHCH2CH2CH2-),2.07(1H,brs,-OH),3.51(1H,t,-OCH2CH<),3.64(2H,t,-CH2OH),4.04(1H,dd,-OCH2CH<),4.07-4.10(1H,m,-OCH2CH<)
(实施例2)
向配备有温度计、氮气进气管、搅拌器和冷凝管的500mL四颈烧瓶中注入式(22)的化合物(20.0g,0.115mol)、三乙胺(23.3g,0.230mol)和甲苯(200g),将混合物冷却至10℃以下。在持续冷却的同时,向其中逐滴加入在滴液漏斗中的甲磺酰氯(19.8g,0.173mol)。在逐滴加入完成之后,在20℃进行反应2小时。加入乙醇(7.97g,0.173mol)并将混合物搅拌一段时间,然后过滤。用离子交换水清洗有机层,用无水硫酸钠干燥,过滤后,在减压下蒸馏去除溶剂,以获得式(23)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.35(3H,s,-CH3),1.40(3H,s,-CH3),1.44-1.83(6H,m,>CHCH2CH2CH2-),3.01(3H,s,-OSO2CH3),3.51(1H,t,-OCH2CH<),4.03-4.11(2H,m,-OCH2CH<,-OCH2CH<),4.24(2H,t,-CH2OSO2CH3)
(实施例3)
向配备有温度计、氮气进气管、搅拌器和冷凝管的500mL四颈烧瓶中注入式(23)的化合物(20.0g,79.3mmol)、邻苯二甲酰亚胺钾盐(17.6g,95.2mmol)和脱水二甲基甲酰胺(200g),在60℃进行反应2小时。将混合物冷却至10℃以下,向其中加入离子交换水(400g)然后搅拌一段时间,用乙酸乙酯/己烷(60/40v/v)的混合溶液萃取该混合物。用0.2重量%的碳酸钾水溶液清洗有机层,并且用无水硫酸钠干燥。过滤后,减压蒸馏去除溶剂,以获得式(24)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.34(3H,s,-CH3),1.39(3H,s,-CH3),1.44-1.75(6H,m,>CHCH2CH2CH2-),3.50(1H,t,-OCH2CH<),3.69(2H,t,-CH2-邻苯二甲酰亚胺),4.01-4.09(2H,m,-OCH2CH<,-OCH2CH<),7.71-7.85(4H,m,-邻苯二甲酰亚胺)
(实施例4)
向配备有温度计、氮气进气管、搅拌器和冷凝管的1L四颈烧瓶中注入式(24)的化合物(15.2g,50.0mmol)、对甲苯磺酸一水合物(951mg,5.00mmol)和甲醇(500mL),在室温下进行反应4小时。向其中加入三乙胺(1.01g,10.0mmol)然后搅拌一段时间,减压蒸馏去除溶剂。将残留物溶解于氯仿,溶液用离子交换水清洗,并且将有机层用无水硫酸钠干燥。过滤后,减压蒸馏去除溶剂,以获得式(25)的化合物。
1H-NMR(CD3CN,内标物TMS);δ(ppm):
1.24-1.61(6H,m,>CHCH2CH2CH2-),2.69(1H,t,-OH),2.75(1H,d,-OH),3.17-3.21(1H,m,-OCH2CH<),3.31-3.37(1H,m,-OCH2CH<),3.39-3.43(1H,m,-OCH2CH<),3.54(2H,t,-CH2-邻苯二甲酰亚胺),7.67-7.75(4H,m,-邻苯二甲酰亚胺)
(实施例5)
向配备有温度计、氮气进气管、搅拌器、Dean-stark管和冷凝管的300mL三颈烧瓶中注入式(25)的化合物(3.87g,14.7mmol)、4-羟基苯甲醛(1.20g,9.83mmol)、吡啶对甲苯磺酸盐(247mg,0.983mmol)和甲苯(180g),进行反应4小时,同时通过与甲苯共沸蒸馏去除副产物水。向其中加入三乙胺(199mg,1.97mmol)然后搅拌一段时间,减压蒸馏去除溶剂。将残留物溶解于氯仿,溶液依次用20重量%氯化钠溶液和离子交换水清洗,并且将有机层用无水硫酸钠干燥。过滤后,减压蒸馏去除溶剂,以获得式(26)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.41-1.80(6H,m,>CHCH2CH2CH2-),3.57-4.26(5H,m,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.71(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.79-6.82(2H,m,芳环H),7.31-7.35(2H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
(实施例6)
向配备有温度计、氮气进气管、搅拌器和冷凝管的300mL四颈烧瓶中注入脱水甲醇(12.8g,0.400mol),脱水甲苯(150g)和金属钠(0.3g,13mmol),在向混合物中鼓入氮气的同时在室温下搅拌混合物直到金属钠溶解。将溶液注入5L高压釜中,并且在用氮气置换系统内部后,将温度升高至100℃。在1MPa以下的压力下,在100至130℃下加入氧化乙烯(1,987g,45mol)之后,再持续反应2小时。在减压下去除未反应的氧化乙烯气体之后,将混合物冷却至60℃并且用85%磷酸溶液将pH调整至7.5,从而获得式(27)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
2.68(1H,t,OH),3.38(3H,s,CH3O-),3.49-3.85(450H,m,-(OCH2CH2)n-)
GPC分析;数量平均分子量(Mn):5119,重量平均分子量(Mw):5226,多分散度(Mw/Mn):1.021
CH3-(OCH2CH2)n-OH (27)
n=约113
(实施例7)
向配备有温度计、氮气进气管、搅拌器、Dean-stark管和冷凝管的500mL三颈烧瓶中注入式(27)的化合物(100g,20.0mmol)和甲苯(250g),并且通过与甲苯共沸蒸馏去除水。在冷却至40℃后,注入三乙胺(3.24g,32.0mmol),向其中逐滴加入在滴液漏斗中制备的甲磺酰氯(2.75g,24.0mmol)。在逐滴加入完成之后,在40℃进行反应3小时。向其中加入乙醇(1.11g,24.0mmol)并将混合物搅拌一段时间,过滤,并且用乙酸乙酯(200g)稀释。通过加入己烷(500g)进行结晶,过滤后,将晶体溶解于乙酸乙酯(500g)。通过加入己烷(500g)再次进行结晶,过滤后,在减压下干燥晶体,以获得式(28)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
3.08(3H,s,-OSO2CH3),3.38(3H,s,CH3O-),3.52-3.85(448H,m,-(OCH2CH2)n-OCH2-),4.37-4.39(2H,m,-CH2OSO2CH3)
GPC分析;数量平均分子量(Mn):5197,重量平均分子量(Mw):5306,多分散度(Mw/Mn):1.021
n=约113
(实施例8)
向配备有温度计、氮气进气管、搅拌器和冷凝管的100mL三颈烧瓶中注入式(28)的化合物(5.00g,1.00mmol)、式(26)的化合物(551mg,1.50mmol)、碳酸钾(691mg,5.00mmol)和乙腈(25g),在80℃进行反应4小时。减压蒸馏去除溶剂之后,将残留物溶解于乙酸乙酯(100g),并过滤溶液。通过加入己烷(100g)进行结晶,过滤后,在减压下干燥晶体,以获得式(29)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),3.38(3H,s,CH3O-),3.52-4.25(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.89-6.91(2H,m,芳环H),7.35-7.39(2H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):5462,重量平均分子量(Mw):5582,多分散度(Mw/Mn):1.022
n=约113
(实施例9)
向配备有温度计、氮气进气管、搅拌器和冷凝管的50mL三颈烧瓶中注入式(29)的化合物(2.00g,0.400mmol)、甲醇(7g)和乙二胺一水合物(0.781g,10.0mmol),在40℃进行反应4小时。用20重量%的氯化钠溶液稀释混合物,用二氯甲烷萃取,并且在减压下蒸馏去除溶剂。将残留物溶解于乙酸乙酯(50g),用无水硫酸钠干燥,过滤,并且通过加入己烷(50g)结晶。过滤后,在减压下干燥晶体,以获得式(30)的化合物。
1H-NMR(CD3OD,内标物TMS);δ(ppm):
1.43-1.79(6H,m,>CHCH2CH2CH2-),2.77(2H,t,-CH2-NH2),3.36(3H,s,CH3O-),3.50-4.29(453H,m,-(OCH2CH2)n-,-OCH2CH<),5.70(0.6H,s,>CH-),5.81(0.4H,s,>CH-),6.93-6.98(2H,m,芳环H),7.33-7.41(2H,m,芳环H)
GPC分析;数量平均分子量(Mn):5332,重量平均分子量(Mw):5454,多分散度(Mw/Mn):1.023
n=约113
(实施例10)
向配备有温度计、氮气进气管、搅拌器和冷凝管的50mL三颈烧瓶中注入式(30)的化合物(0.20g,0.040mmol)和乙腈(10g),向其中加入N-琥珀酰亚胺基-3-马来酰亚胺基丙酸酯(32mg,0.048mmol),在25℃进行反应3小时。过滤后,减压蒸馏去除溶剂。将残留物溶解于乙酸乙酯(25g),并且通过加入己烷(25g)结晶。过滤后,在减压下干燥晶体,以获得式(31)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),2.44(2H,t,-CH2CH2-马来酰亚胺),3.27-3.37(2H,m,-CH2NHCO-),3.38(3H,s,CH3O-),3.47-4.25(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2CH2-马来酰亚胺),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.15(1H,brs,-NHCO-),6.70(2H,s,-马来酰亚胺),6.89-6.91(2H,m,芳环H),7.35-7.39(2H,m,芳环H)
GPC分析;数量平均分子量(Mn):5484,重量平均分子量(Mw):5610,多分散度(Mw/Mn):1.023
n=约113
(实施例11)
使用3-氟-4-羟基苯甲醛,以与实施例1至8相同的方法获得式(32)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(6H,m,>CHCH2CH2CH2-),3.38(3H,s,CH3O-),3.52-4.23(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.95-7.21(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):5485,重量平均分子量(Mw):5606,多分散度(Mw/Mn):1.022
n=约113
(实施例12)
使用2-溴-5-羟基苯甲醛,以与实施例1至8相同的方法获得式(33)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(6H,m,>CHCH2CH2CH2-),3.38(3H,s,CH3O-),3.52-4.23(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.95-7.21(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):5548,重量平均分子量(Mw):5670,多分散度(Mw/Mn):1.022
n=约113
(实施例13)
以与实施例1至4相似的方法合成式(34)的化合物,并且使用3-氟-4-羟基苯甲醛,以与实施例5至8相同的方法获得式(35)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.89(2H,m,-CH2CH2-邻苯二甲酰亚胺),3.19(1H,m,-OCH2CH<),3.38(3H,s,CH3O-),3.52-4.41(456H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2CH2CH2-邻苯二甲酰亚胺),5.34(0.8H,s,>CH-),5.42(0.2H,s,>CH-),6.95-7.25(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):5498,重量平均分子量(Mw):5619,多分散度(Mw/Mn):1.022
n=约113
(实施例14)
使用式(34)的化合物和2-溴-5-羟基苯甲醛,以与实施例5至8相同的方法获得式(36)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.89(2H,m,-CH2CH2-邻苯二甲酰亚胺),3.19(1H,m,-OCH2CH<),3.38(3H,s,CH3O-),3.52-4.41(456H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2CH2CH2-邻苯二甲酰亚胺),5.61(0.8H,s,>CH-),5.68(0.2H,s,>CH-),6.78-7.40(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):5564,重量平均分子量(Mw):5686,多分散度(Mw/Mn):1.022
n=约113
(实施例15)
n=约113
通过使用盐酸去除根据JP-A-2010-248504中描述的方法合成的式(37)的化合物的叔丁基,获得式(38)的化合物。
1H-NMR(D2O,内标物TMS);δ(ppm):
3.14(2H,t,NH2CH2-),3.40-4.00(452H,m,-(OCH2CH2)n-OCH2-)
NH2(CH2)2-(OCH2CH2)n-OH
(38)
n=约113
(实施例16)
通过使式(38)的化合物与5-叠氮戊酸酐反应获得式(39)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.60-1.74(4H,m,-CH2CH2CH2CH2N3),2.18(2H,t,-CH2CH2CH2CH2N3),3.29(2H,t,-CH2CH2CH2CH2N3),3.40-3.85(454H,m,-(OCH2CH2)n-OCH2-,-CONHCH2-),6.30(1H,brs,-CONH-)
n=约113
(实施例17)
以与实施例7相似的方法,通过在三乙胺的存在下,使式(39)的化合物与甲磺酰氯在甲苯中反应,获得式(40)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.60-1.74(4H,m,-CH2CH2CH2CH2N3),2.18(2H,t,-CH2CH2CH2CH2N3),3.08(3H,s,-OSO2CH3),3.29(2H,t,-CH2CH2CH2CH2N3),3.40-3.85(452H,m,-(OCH2CH2)n-OCH2-,-CONHCH2-),4.37-4.39(2H,m,-CH2OSO2CH3),6.30(1H,brs,-CH2CONH-)
n=约113
(实施例18)
使用3-氟-4-羟基苯甲醛和式(40)的化合物,以与实施例1至5和8相同的方法获得式(41)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(10H,m,>CHCH2CH2CH2-,-CH2CH2CH2CH2N3),2.18(2H,t,-CH2CH2CH2CH2N3),3.28-4.23(461H,m,-(OCH2CH2)n-OCH2-,-CH2CH2CH2CH2N3,-CH2CONHCH2-,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.30(1H,brs,-CH2CONH-),6.95-7.21(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
n=约113
(实施例19)
通过与实施例9相同的方式从式(41)的化合物去保护邻苯二甲酰亚胺基,随后与碘代乙酸酐反应,获得式(42)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(10H,m,>CHCH2CH2CH2-,-CH2CH2CH2CH2N3),2.18(2H,t,-CH2CH2CH2CH2N3),3.28-4.23(463H,m,-(OCH2CH2)n-OCH2-,-CH2CH2CH2CH2N3,-CH2CONHCH2-,ICH2CONHCH2-,ICH2CONHCH2-,-OCH2CH<),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.30(1H,brs,-CH2CONH-),6.96(1H,brs,ICH2CONHCH2-),6.95-7.21(3H,m,芳环H)
GPC分析;数量平均分子量(Mn):5679,重量平均分子量(Mw):5815,多分散度(Mw/Mn):1.024
n=约113
(实施例20)
n=约113
以与实施例7相同的方法,使根据JP-A-2004-197077中描述的方法合成的式(43)的化合物与甲磺酰氯反应,获得式(44)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
3.08(3H,s,-OSO2CH3),3.38(6H,s,CH3O-),3.40-4.00(903H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<),4.26-4.42(2H,m,-CH2OSO2CH3)
n=约113
(实施例21)
使用3-氟-4-羟基苯甲醛和式(44)的化合物,以与实施例1至5和8相同的方法获得式(45)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(6H,m,>CHCH2CH2CH2-),3.38(6H,s,CH3O-),3.40-4.23(910H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.95-7.21(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):9761,重量平均分子量(Mw):9986,多分散度(Mw/Mn):1.023
n=约113
(实施例22)
n=约113
在三乙胺和4-二甲基氨基吡啶的存在下,使根据JP-A-2004-197077中描述的方法合成的式(46)的化合物与乙酸酐反应,获得式(47)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
2.08(6H,s,CH3CO-),3.40-4.00(901H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<,-CH2OCH2Ph),4.22(4H,t,CH3CO2CH2-),4.54(2H,s,-CH2OCH2Ph),7.27-7.38(5H,m,-CH2OCH2Ph)
n=约113
(实施例23)
根据JP-A-2004-197077中描述的方法从式(47)的化合物中去除苯甲基,随后以与实施例7相似的方法使其与甲磺酰氯反应,获得式(48)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
2.08(6H,s,CH3CO-),3.08(3H,s,-OSO2CH3),3.40-4.00(899H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<),4.22(4H,t,CH3CO2CH2-),4.26-4.42(2H,m,-CH2OSO2CH3)
n=约113
(实施例24)
使用3-氟-4-羟基苯甲醛和式(48)的化合物,以与实施例1至5和8相同的方法获得式(49)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(6H,m,>CHCH2CH2CH2-),2.08(6H,s,CH3CO-),3.40-4.23(910H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<,-OCH2CH<,-CH2-邻苯二甲酰亚胺,CH3CO2CH2-),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.95-7.21(3H,m,芳环H),7.70-7.86(4H,m,-邻苯二甲酰亚胺)
n=约113
(实施例25)
通过使用乙二胺一水合物从式(49)的化合物去保护邻苯二甲酰亚胺基并使用氢氧化钠水溶液去除乙酰基,随后以与实施例10相同的方式使其与N-琥珀酰亚胺基-3-马来酰亚胺基丙酸酯反应,获得式(50)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),2.44(2H,t,-CH2CH2-马来酰亚胺),3.27-3.37(2H,m,-CH2NHCO-),3.40-4.23(910H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<,-OCH2CH<,-CH2CH2-马来酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.15(1H,brs,-NHCO-),6.70(2H,s,-马来酰亚胺),6.95-7.21(3H,m,芳环H)
n=约113
(实施例26)
在三乙胺的存在下,使式(50)的化合物与N,N'-二琥珀酰亚胺基碳酸酯在二氯甲烷中反应,获得式(51)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),2.44(2H,t,-CH2CH2-马来酰亚胺),2.84(8H,s,-琥珀酰亚胺),3.27-3.37(2H,m,-CH2NHCO-),3.40-4.23(906H,m,-(OCH2CH2)n-OCH2-,-(OCH2CH2)n-OCH<,-OCH2CH<,-CH2CH2-马来酰亚胺),4.44-4.48(4H,m,-CH2O-COO-琥珀酰亚胺),5.70(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.15(1H,brs,-NHCO-),6.70(2H,s,-马来酰亚胺),6.95-7.21(3H,m,芳环H)
GPC分析;数量平均分子量(Mn):9955,重量平均分子量(Mw):10204,多分散度(Mw/Mn):1.025
n=约113
(实施例27)
n=约113
以与实施例7相同的方法,使通过将氧化乙烯聚合至季戊四醇而合成的式(52)的化合物与甲磺酰氯反应,获得式(53)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
3.08(12H,s,-OSO2CH3),3.47-3.85(1800H,m,-(OCH2CH2)n-OCH2-),4.37-4.39(8H,m,-CH2OSO2CH3)
n=约113
(实施例27)
使用3-氟-4-羟基苯甲醛和式(53)的化合物,以与实施例1至5和8相同的方法获得式(54)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.38-1.80(24H,m,>CHCH2CH2CH2-),3.52-4.23(1828H,m,-(OCH2CH2)n-OCH2-,-OCH2CH<,-CH2-邻苯二甲酰亚胺),5.70(2.4H,s,>CH-),5.82(1.6H,s,>CH-),6.95-7.21(12H,m,芳环H),7.70-7.86(16H,m,-邻苯二甲酰亚胺)
GPC分析;数量平均分子量(Mn):19645,重量平均分子量(Mw):20136,多分散度(Mw/Mn):1.025
n=约113
(实施例28)
将式(29)、式(32)、式(33)、式(35)和式(36)的各个化合物(20mg)分别溶解于pD 5.5的MES氘代水缓冲剂(1mL)和pD 7.4的HEPES氘代水缓冲剂(1mL)中,使其静置于37℃的恒温水浴中。图1和图2分别示出在pD 5.5和pD 7.4时水解率的测量结果。
如图1所示,式(29)、式(32)、式(33)、式(35)和式(36)的化合物在pD 5.5、37℃下的水解率半衰期(t1/2)分别为2小时、12小时、30天、24小时和6个月。另外,如图2所示,在pD 7.4和37℃下,式(29)和式(32)的化合物的水解率半衰期(t1/2)分别为65小时和18天,对于式(35)的化合物在18天时观察到大约17%的水解,并且对于式(33)和式(36)的化合物甚至在18天后也没有观察到水解。
(实施例29)
向配备有温度计、氮气进气管、搅拌器和冷凝管的100mL三颈烧瓶中注入式(26)的化合物(1.10g,3.00mmol)、甲醇(53g)和乙二胺一水合物(5.86g,75.0mmol),在40℃进行反应4小时。将混合物冷却至室温,用10重量%的氯化钠水溶液稀释,并且用二氯甲烷萃取。用10重量%的氯化钠溶液清洗有机层,并且用无水硫酸钠干燥。过滤后,减压蒸馏去除溶剂,以获得式(56)的化合物。
1H-NMR(CD3OD,内标物TMS);δ(ppm):
1.44-1.80(6H,m,>CHCH2CH2CH2-),2.77(2H,t,-CH2-NH2),3.55-4.31(3H,m,-OCH2CH<),5.70(0.6H,s,>CH-),5.81(0.4H,s,>CH-),6.89-6.94(2H,m,芳环H),7.29-7.37(2H,m,芳环H)
(实施例30)
向配备有温度计、氮气进气管和搅拌器的100mL三颈烧瓶中注入式(56)的化合物(593mg,2.50mmol)和脱水甲醇(20g),并且在逐滴加入通过将三氟乙酸乙酯(426mg,3.00mmol)溶解于脱水甲醇(20g)获得的溶液的同时搅拌混合物。在逐滴加入完成之后,在25℃进行反应2小时。减压蒸馏去除溶剂,并且在减压下干燥残留物,以获得式(57)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.44-1.80(6H,m,>CHCH2CH2CH2-),3.57-4.26(5H,m,-OCH2CH<,-CH2-NHCOCF3),5.71(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.79-6.82(2H,m,芳环H),7.31-7.35(3H,m,芳环H,-NHCOCF3)
(实施例31)
n=约113
以与实施例7相同的方法,使根据JP-A-2010-138388中描述的方法合成的式(58)的化合物与甲磺酰氯反应,随后使用盐酸去除叔丁基,获得式(59)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
2.68(1H,t,-OH),3.08(3H,s,-OSO2CH3),3.52-3.85(448H,m,-(OCH2CH2)n-OCH2-),4.37-4.39(2H,m,-CH2OSO2CH3)
GPC分析;数量平均分子量(Mn):5195,重量平均分子量(Mw):5309,多分散度(Mw/Mn):1.022
(实施例32)
n=约113
向配备有温度计、氮气进气管、搅拌器和冷凝管的100mL三颈烧瓶中注入式(59)的化合物(5.00g,1.00mmol)、式(57)的化合物(500mg,1.50mmol)、碳酸钾(691mg,5.00mmol)和乙腈(25g),在80℃进行反应4小时。减压蒸馏去除溶剂之后,将残留物溶解于乙酸乙酯(100g),并过滤溶液。通过加入己烷(100g)进行结晶,过滤后,在减压下干燥晶体,以获得式(60)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),2.68(1H,t,-OH),3.52-4.25(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2-NHCOCF3),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.89-6.91(2H,m,芳环H),7.31-7.39(3H,m,芳环H,-NHCOCF3)
GPC分析;数量平均分子量(Mn):5432,重量平均分子量(Mw):5552,多分散度(Mw/Mn):1.022
n=约113
(实施例33)
向配备有温度计、氮气进气管和搅拌器的100mL三颈烧瓶中注入式(60)的化合物(1.00g,0.200mmol)和二氯甲烷(5g),向其中加入戊二酸酐(34.2mg,0.300mmol)、三乙胺(30.4mg,0.300mmol)和4-二甲基氨基吡啶(1.2mg,0.015mmol),在25℃进行反应6小时。过滤后,减压蒸馏去除溶剂。将残留物溶解于甲醇(2.5g),向其中加入1M碳酸钾水溶液(5g),并且在25℃进行反应3小时。用20重量%的氯化钠水溶液稀释混合物,用二氯甲烷萃取,并且在减压下蒸馏去除溶剂。将残留物溶解于乙酸乙酯(50g),用无水硫酸钠干燥,过滤,并且通过加入己烷(50g)结晶。过滤后,在减压下干燥晶体,以获得式(61)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),1.97(2H,quin,-CH2CH2CH2COOH),2.38-2.46(4H,m,-CH2CH2CH2COOH),2.71(2H,t,-CH2-NH2),3.52-4.27(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CHbO-COCH2-),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.89-6.91(2H,m,芳环H),7.35-7.39(2H,m,芳环H)
GPC分析;数量平均分子量(Mn):5449,重量平均分子量(Mw):5569,多分散度(Mw/Mn):1.022
n=约113
(实施例34)
向配备有温度计、氮气进气管、搅拌器、Dean-stark管和冷凝管的300mL三颈烧瓶中注入1,2,6-己三醇(2.01g,15.0mmol)、4-羟基苯甲醛(1.22g,10.0mmol)、对甲苯磺酸一水合物(19.0mg,0.100mmol)和甲苯(183g),进行反应4小时,同时通过与甲苯共沸蒸馏去除副产物水。向其中加入三乙胺(20.2mg,0.200mmol)然后搅拌一段时间,用10重量%的氯化钠溶液清洗混合物,并且用无水硫酸钠干燥有机层。过滤后,减压蒸馏去除溶剂,以获得式(62)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.42-1.80(6H,m,>CHCH2CH2CH2-),3.57-4.26(5H,m,-OCH2CH<,-CH2-OH),5.71(0.6H,s,>CH-),5.82(0.4H,s,>CH-),6.79-6.82(2H,m,芳环H),7.31-7.35(2H,m,芳环H)
(实施例35)
向配备有温度计、氮气进气管、搅拌器和冷凝管的100mL三颈烧瓶中注入式(28)的化合物(5.00g,1.00mmol)、式(62)的化合物(357mg,1.50mmol)、碳酸钾(691mg,5.00mmol)和乙腈(25g),在80℃进行反应4小时。减压蒸馏去除溶剂之后,将残留物溶解于乙酸乙酯(100g),并过滤溶液。通过加入己烷(100g)进行结晶,过滤后,在减压下干燥晶体,以获得式(63)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.40-1.81(6H,m,>CHCH2CH2CH2-),3.38(3H,s,CH3O-),3.52-4.25(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2-OH),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.89-6.91(2H,m,芳环H),7.35-7.39(2H,m,芳环H)
GPC分析;数量平均分子量(Mn):5332,重量平均分子量(Mw):5449,多分散度(Mw/Mn):1.022
n=约113
(实施例36)
在三乙胺的存在下,使式(63)的化合物与N,N'-二琥珀酰亚胺基碳酸酯在二氯甲烷中反应,获得式(64)的化合物。
1H-NMR(CDCl3,内标物TMS);δ(ppm):
1.45-1.87(6H,m,>CHCH2CH2CH2-),2.84(4H,两个单峰(two singlets),-琥珀酰亚胺),3.38(3H,s,CHbO-),3.52-4.38(455H,m,-(OCH2CH2)n-,-OCH2CH<,-CH2O-COO-琥珀酰亚胺),5.72(0.6H,s,>CH-),5.84(0.4H,s,>CH-),6.89-6.91(2H,m,芳环H),7.35-7.39(2H,m,芳环H)
GPC分析;数量平均分子量(Mn):5470,重量平均分子量(Mw):5590,多分散度(Mw/Mn):1.022
n=约113
尽管已详细地并参考具体实施方式对本发明进行了描述,但对于本领域技术人员来说,显然可以在不背离本发明的精神和范围时,在其中做出各种改变和修改。
本发明基于2014年3月31日提交的日本专利申请号2014-72356和2014年9月22日提交的日本专利申请号2014-193039,其全部内容通过引用并入本文。而且,本文中引用的所有参考文献以其整体并入本文。
- 还没有人留言评论。精彩留言会获得点赞!