通过在掺硼金刚石阳极上使未活化或去活化的芳族体系直接电化学胺化以制备1,5‑二氨基萘和1‑氨基‑5‑硝基萘的方法与流程
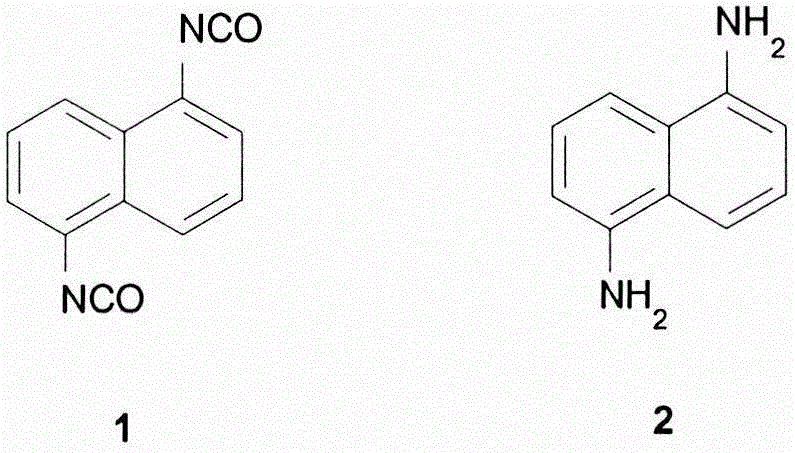
1,5-二氨基萘(2)特别是用于制备1,5-萘二异氰酸酯(1)的重要中间体(图1)。此芳族二异氰酸酯是用于高强度聚氨酯的重要的基础结构单元。
1,5-萘二异氰酸酯的工业合成起始自转化为1,5-萘二磺酸(4)的萘(3)。1,5-萘二磺酸在碱熔体中转化为1,5-二羟基萘(5),其接着在布赫尔反应中转化为二胺2。随后进行光气化以得到1,5-萘二异氰酸酯(1)。1,5-萘二异氰酸酯的此经典合成示于图示1中。
这里描述的工艺的严重缺点是在萘的磺化时形成异构体混合物。除了所希望的1,5-化合物之外,还得到另外的二磺酸和三磺酸。这伴随着基于使用的萘计的高收率损失。在形成二羟基化合物的反应中严苛且腐蚀性的条件代表了所述经典工艺的另外的缺陷。所述多个合成步骤伴随着耗时的中间体后处理。此外,由于多个工艺阶段而产生高的能源成本。日渐紧缺的原料和环境保护的上升的重要性因此要求用于制备1,5-二氨基萘(2)的新的合成策略。
本发明的目的是提供环保且同时廉价地得到在1,5位二取代的萘衍生物,特别是1,5-二氨基萘的途径。高强度聚氨酯的重要前体,即1,5-二氨基萘的竞争性合成制备可导致这些坚固且耐用的聚合物更广泛的使用。
本发明提供一种使用掺硼金刚石阳极使未活化或去活化的芳族体系进行氧化性电化学胺化的方法。
去活化的芳族体系被理解为是指其环中的电子密度由于取代基的诱导效应和/或中介效应而降低的取代的芳族化合物。未活化的芳族体系被理解为是指未取代的芳族化合物。如果取代的芳族化合物既具有由于诱导效应和/或中介效应降低环中的电子密度的取代基也具有由于诱导效应和/或中介效应增加环中的电子密度的取代基,当环中的电子密度整体降低时,该类芳族化合物属于根据本发明定义的去活化芳族体系。
在本发明的方法中,使用萘、在1位取代的萘衍生物以及多环的稠合芳族化合物(≥3环)和二环或多环的且在环中具有至少一个氮原子或氧原子的稠合杂芳族化合物作为去活化或贫电子芳族体系。在1位取代的萘衍生物包括,例如,1-硝基萘、1-氰基萘和1-烷氧基羰基萘或芳氧基羰基萘。稠合芳族化合物的实例是蒽、菲、、芘、苝和蔻。杂芳族化合物的实例是喹啉、异喹啉、酞嗪、喹唑啉、萘啶、吖啶、吩嗪、菲啶和菲咯啉,例如1,7-或1,10-菲咯啉。
优选萘和在1位取代的萘衍生物,特别优选萘和1-硝基萘。
本发明提供一种在掺硼金刚石阳极上使
a)1-硝基萘、1-氰基萘、1-烷氧基羰基萘或1-芳氧基羰基萘在5位,或
b)萘在1和5位
进行氧化性电化学胺化的方法。
优选1-硝基萘在5位或萘在1和5位的胺化。
可考虑的胺化试剂尤其是吡啶及其取代和稠合的衍生物如甲基吡啶(2-、3-和4-甲基吡啶)、二甲基吡啶(2,3-、2,4-、2,5-、2,6-、3,4-和3,5-二甲基吡啶)和三甲基吡啶(2,3,4-、2,3,5-、2,3,6-、2,4,5-、2,4,6-和3,4,5-三甲基吡啶)、具有混合的烷基取代基的吡啶,例如5-乙基二甲基吡啶及其异构体,以及喹啉和异喹啉。
本发明提供一种使用掺硼金刚石阳极使用吡啶、一种或多种甲基吡啶异构体、一种或多种二甲基吡啶异构体、一种或多种三甲基吡啶异构体、喹啉、异喹啉或这些化合物的混合物作为胺化试剂使未活化或去活化的芳族体系进行氧化性电化学胺化的方法。
本发明同样提供一种使用掺硼金刚石阳极使用吡啶、一种或多种甲基吡啶异构体、一种或多种二甲基吡啶异构体、一种或多种三甲基吡啶异构体、一种或多种具有混合的烷基取代基的吡啶异构体、喹啉、异喹啉或这些化合物的混合物,优选5-乙基二甲基吡啶和/或一种或多种其异构体作为胺化试剂使未活化或去活化的芳族体系进行氧化性电化学胺化的方法。
本发明提供一种在硼掺杂的阳极上使用吡啶、一种或多种甲基吡啶异构体、一种或多种二甲基吡啶异构体、一种或多种三甲基吡啶异构体、喹啉、异喹啉或这些化合物的混合物作为胺化试剂使
a)1-硝基萘、1-氰基萘、1-烷氧基羰基萘或1-芳氧基羰基萘在5位,或
b)萘在1和5位
进行电化学胺化的方法。
优选的胺化试剂是吡啶。
一系列带有质子的亲核试剂对于随后的使(一个或多个)伯氨基团游离(Freisetzung)的反应是原则上合适的,可以提及的是水、氢氧化物、氢过氧化物、氨、酰胺、肼、酰肼、羟胺、式NH2R1和NHR1R2的伯胺或仲胺,其中R1和R2为具有1-10个碳原子且任选地含有选自氧、硫、氮的杂原子的线性或支化的、饱和或不饱和的、脂族、芳脂族、环脂族或芳族基团,或者在NHR1R2的情况下共同形成具有1-6个碳原子且任选地含有选自氧、硫和氮的杂原子的饱和或不饱和的环。
优选氢氧化物、氨、肼、羟胺和哌啶,特别优选哌啶。
本发明还提供一种使用掺硼金刚石阳极使未活化或去活化的芳族体系进行氧化性电化学胺化并随后由伯胺化产物使胺游离的方法。
本发明还提供一种通过在掺硼金刚石阳极上使1-硝基萘、1-氰基萘、1-烷氧基羰基萘或1-芳氧基羰基萘在5位进行氧化性电化学胺化并随后由伯胺化产物使胺游离以制备1-氨基-5-硝基萘、1-氨基-5-氰基萘、1-氨基-5-烷氧基羰基萘或1-氨基-5-芳氧基羰基萘的方法。
优选通过此方法由1-硝基萘制备1-氨基-5-硝基萘,以及通过此路线制备的1-氨基-5-硝基萘本身。
本发明还提供一种通过在掺硼金刚石阳极上使萘在1和5位进行氧化性电化学胺化并随后由伯胺化产物使胺游离以制备1,5-二氨基萘的方法。
本发明同样提供所描述的最后两种的方法,其中使用吡啶用于电化学胺化并使用哌啶用于由伯胺化产物使胺游离。
形成二胺2的经典的多阶段合成法通过1-硝基萘在5位的直接电化学胺化和萘在1,5位的双胺化大大缩短。另外,可以避免经典方法中在萘的1,5-官能化时由于缺乏区域选择性和/或形成三取代和更高取代的中间产物而发生的收率损失。
富电子芳族体系使用石墨毡作为电极材料的电化学胺化已经被Morofuji等在2013年发表(T.Morofuji,A.Shimizu,J.-I.Yoshida,J.Am.Chem.Soc.2013,135,5000-5003)。与此相反,在本发明中,贫电子,即未活化或甚至去活化的芳族化合物的电化学胺化发生在由掺硼金刚石(BDD)制成的阳极上。贫电子起始物质在其它阳极材料(石墨、玻璃碳、铂)上的反应是不可能的。
此外,文献中描述了使用Ti(IV)/Ti(III)作为氧化还原介体和羟胺作为氮源使茴香醚在硫酸/乙腈中进行电化学胺化(Y.A.Lisitsin,L.V.Grigor’eva,Russ.J.Gen.Chem.2008,78,1009-1010,Y.A.Lisitsyn,L.V.Grigor’eva,Russ.J.Electrochem.2009,45,132-138,Y.A.Lisitsyn,A.V.Sukhov,Russ.J.Electrochem.2011,47,1180-1185,Y.A.Lisitsyn,A.V.Sukhov,Russ.J.Phys.Chem.2012,86,1033-1034,Y.A.Lisitsyn,A.V.Sukhov,Russ.J.Electrochem.2013,49,91-95,Y.A.Lisitsyn,A.V.Sukhov,Russ.J.Gen.Chem.2013,83,1457-1458)。
在所述文献中同样已知由相应的芳族硝基化合物进行硝基苯胺的电化学合成。这里,在氧化之前的反应步骤中发生合适的氮亲核试剂向贫电子的硝基芳族化合物的亲核攻击。作为中间体形成的迈森海默配合物的氧化最终得到取代的芳族硝基化合物。1-硝基萘通过此方法在2位胺化,1,3-二硝基萘在4位胺化。因此,仅仅环A的官能化是可能的(H.Cruz,I.Gallardo,G.Guirado,Green Chem.2011,13,2531-2542,I.Gallardo,G.Guirado,J.Marquet,Eur.J.Org.Chem.2002,2002,251-259)。
根据本发明的1-硝基萘6的电化学胺化通过在BDD阳极上作为中间体形成自由基阳离子来进行,其被作为亲核试剂的吡啶或其上面提及的衍生物捕获(在图示2中,以吡啶为例示出)。作为溶剂,可以使用例如乙腈。由于1-硝基萘的A环比B环更强烈地去活化,吡啶的攻击优选在5位发生(图示2)。在此位置上,取代基也位于彼此的最大距离。由伯萘基吡啶鎓体系7使胺游离最终得到1-氨基-5-硝基萘8(图示2),其可以催化氢化形成所需要的二胺2。
另外,在萘的1,5位选择性引入两个氨基官能的问题通过萘的双电化学胺化得以解决。在BBD阳极上作为中间体形成的自由基阳离子被作为亲核试剂的吡啶或其上面提及的衍生物捕获(在图示3中,以吡啶为例示出)。作为溶剂,可以使用例如乙腈。因为最初形成的简单的1-萘基吡啶鎓中间体在1位已存在正电荷,第二个吡啶分子或其上面提及的衍生物由于静电原因在5位攻击(在图示3中,以吡啶为例示出)。1,5-萘基二吡啶鎓化合物7通过哌啶的氨解得到所需要的二胺2(图示3)。
通过此直接的胺化方法,开启对现存的多阶段合成途径的廉价且环保的替代方案。
实施例
图1:1,5-萘二异氰酸酯(1)和1,5-二-氨基萘(2)。
图2:在屏蔽块中分隔的特氟隆电池(左侧),带有BDD电极和磁力搅拌棒的分隔的H电池(右侧);电极尺寸:分别为10×70mm;阳极空间与阴极空间的分离分别通过由孔隙率为4的烧结玻璃制成且直径为10mm的多孔隔膜实现;溶剂体积:分别为6ml;电极间距:250mm(特氟隆电池),350mm(H电池)。
图3:1-氨基-5-硝基萘(8)的晶体结构。
色谱法:
通过“快速色谱法”的制备型液体色谱分离使用1.6bar的最大压力在公司Macherey-Nagel GmbH&Co,D üren的硅胶60M(0.040-0.063mm)上进行。不加压的分离在公司Merck KGaA,Darmstadt的硅胶Geduran Si 60(0.063-0.200mm)上进行。用作洗脱剂的溶剂(乙酸乙酯(工业级)、环己烷(工业级))预先通过在旋转蒸发器上蒸馏进行纯化。
对于薄层色谱法(DC)使用公司Merck KGaA,Darmstadt的PSC即用板硅胶60F254进行。根据使用的洗脱剂混合物给出Rf值(滞留因数)。为了使DC板着色,使用铈-钼磷酸溶液作为漫渍试剂。铈-钼磷酸试剂:在200ml水中5.6g钼磷酸、2.2g四水合硫酸铈(IV)和13.3g浓硫酸。
气相色谱法(GC/GCMS):
通过公司Shimadzu,日本的气相色谱仪GC-2010对产物混合物和纯物质进行气相色谱检测(GC)。在公司Agilent Technologies,USA的石英毛细管柱HP-5(长度:30m;内径:0.25mm;共价结合的固定相的膜厚度:0.25μm;载气:氢气;注射器温度:250℃;检测器温度:310℃;程序:“硬(hart)”法:50℃初始温度1分钟,加热速率:15℃/分钟,290℃最终温度8分钟)上进行测量。通过气相色谱仪GC-2010与公司Shimadzu,日本的质量检测器GCMS-QP2010的联合记录产物混合物与纯物质的气相色谱质谱(GCMS)。在公司Agilent Technologies,USA的石英毛细管柱HP-1(长度:30m;内径:0.25mm;共价结合的固定相的膜厚度:0.25μm;载气:氢气;注射器温度:250℃;检测器温度:310℃;程序:“硬”法:50℃初始温度1分钟,加热速率:15℃/min,290℃最终温度8分钟;GCMS:离子源的温度:200℃)上进行测量。
质谱法:
所有的电喷雾电离测量(ESI+)都在公司Waters Micromasses,Milford,Massachusetts的QTof Ultima 3上进行。
NMR波谱法:
NMR-波谱检测在公司Bruker,Analytische Messtechnik,Karlsruhe的AC 300或AV II 400型多核共振波谱仪上进行。使用d6-DMSO作为溶剂。1H和13C谱按照公司Cambridge Isotopes Laboratories,USA的NMR Solvent Data Chart根据未氘代溶剂的残余含量进行校准。1H和13C信号的赋值借助于H,H-COSY(相关谱)、H,C-HSQC(异核单量子相干谱)和H,C-HMBC谱(异核多键相关谱)进行。化学位移以ppm为单位的δ值示出。对于NMR信号的多重性使用下面的缩写:s(单峰),bs(宽的单峰),d(双重峰),t(三重峰),q(四重峰),m(多重峰),dd(双重峰的双重峰),dt(三重峰的双重峰),tq(四重峰的三重峰)。所有的耦合常数J都用包括的键数以赫兹(Hz)示出。信号赋值时所示的编号对应于公式图示中给出的编号,其不必与IUPAC命名法一致。
单晶结构分析:
单晶结构分析在Mainz的Johannes Gutenberg大学的有机化学学院在公司STOE&Cie GmbH,Darmstadt的IPDS 2T型仪器上进行。
电化学胺化的通用操作规程:
电化学反应在分隔的特氟隆电池或H电池中进行(图2)。使用掺硼金刚石作为阳极材料。使用铂作为阴极材料。将由各种芳族化合物(0.1mol×1-1)和吡啶(2.4mol×1-1,干燥)在0.2摩尔浓度Bu4NBF4/乙腈混合物(5ml,干燥)中构成的溶液加入阳极空间中。将三氟甲磺酸(0.4ml)在0.2摩尔浓度Bu4NBF4/乙腈混合物(5ml,干燥)中的溶液加入阴极空间中。电解在60℃下通过恒定电流进行。
在已经达到各自的电荷量之后,将反应溶液转移进100ml圆底烧瓶中并在减压下除去乙腈。随后添加10ml乙腈与1ml哌啶,并将该溶液在80℃下在回流下加热12小时。通过GC、DC和GC/MS对反应混合物检测胺化产物。
图示2显示了在掺硼金刚石阳极上1-硝基萘直接电化学胺化为1-氨基-5-硝基萘。例如选择下面的量、材料和工艺参数:0.5mmol 1-硝基萘;12mmol吡啶;0.2M Bu4NBF4/乙腈;溶剂:乙腈;阳极:BDD(1.5cm2);阴极:铂(1.5cm2);温度:60℃;电荷量:144C;电流密度:j=10mA cm-2。胺的游离:哌啶;溶剂:乙腈。
图示3显示了在掺硼金刚石阳极上萘直接电化学胺化为1,5-二-氨基萘。例如选择下面的量、材料和工艺参数:0.5mmol 1-硝基萘;12mmol吡啶;0.2M Bu4NBF4/乙腈;溶剂:乙腈;阳极:BDD(1.5cm2);阴极:铂(1.5cm2);温度:60℃;电荷量:288C;电流密度:j=10mA cm-2。胺的游离:哌啶;溶剂:乙腈;温度:80℃;反应时间:12小时。
实施例1(根据本发明):1-氨基-5-硝基萘(8)的制备
根据上面的操作规程,将0.09g(0.53mmol,0.04当量)1-硝基萘、0.30g(0.91mmol,0.07当量)四氟硼酸四丁基铵、1ml(0.98g,12.41mmol,1.0当量)吡啶溶解在5ml干燥乙腈中并加入阳极空间中。将0.3g(0.91mmol)四氟硼酸四丁基铵与0.4ml三氟甲烷磺酸在5ml干燥乙腈中的溶液加入阴极空间中。电解在分隔的特氟隆电池中进行。
阳极:BDD;电极面积:1.5cm2。
阴极:铂;电极面积:1.5cm2。
电荷量:144C。
电流密度:j=10mA cm-2。
温度:60℃。
电解时间已经过去之后,在减压下除去溶剂。随后添加10ml乙腈和1ml(0.86g,10.00mmol,0.8当量)哌啶。反应溶液在80℃下在回流下加热12小时。减压除去溶剂并使用公司B üchi-LabortechnikGmbH的分离系统通过柱色谱在硅胶上纯化残余物。使用长度为150mm且直径为40mm的分离柱,并在10bar的最大压力和50ml×min-1的流动速度下操作。这里使用下面的溶剂梯度:首先是环己烷/乙酸乙酯95∶5,所述溶剂梯度随后在第一个40分钟内提高到环己烷/乙酸乙酯3∶1(RF=0.16)。得到19.6mg(0.1mmol,21%)淡红色的固体。
1H-NMR(400MHz,DMSO):δ(ppm)=6.18(s,2H,NH2),6.83(dd,3J=7.2Hz,4J=1.3Hz,1H,H-2),7.35-7.56(m,3H,H-3,4,7),8.14(d,3J=7.5Hz,1H,H-8),8.49(d,3J=8.5Hz,1H,H-6)。
13C-NMR(101MHz,DMSO):δ(ppm)=108.4(C-4),108.8(C-2),121.8,123.37(C-8),125.4(C-4a),128.2(C-6),130.6,146.0(C-1),146.6(C-5)。
GC(硬法,HP-5):tR=12.78min(滞留时间)。
对C10H9N2O2计算的HRMS(高分辨质谱法):189.0664;测得:189.0662。
晶体结构分析(晶体结构:图3):
通过从正庚烷/甲醇使20mg的8结晶得到单晶。
所述化合物的结构数据以SHE1332的命名存放在Mainz大学有机学院(Dr.D.Schollmeyer,X-射线结构分析,Duesbergweg 10-14,55128Mainz)。
实施例2(根据本发明):1,5-二氨基萘(2)的制备
根据通用的操作规程,将0.064g(0.50mmol,0.04当量)萘、0.30g(0.91mmol,0.07当量)四氟硼酸四丁基铵、1ml(0.98g,12.41mmol,1.0当量)吡啶溶解在5ml干燥乙腈中并加入阳极空间中。将0.3g(0.91mmol)四氟硼酸四丁基铵与0.4ml三氟甲磺酸在5ml干燥乙腈中的溶液加入阴极空间中。电解在分隔的H电池中进行。
阳极:BDD;电极面积:1.5cm2。
阴极:铂;电极面积:1.5cm2。
电荷量:288C。
电流密度:j=10mA cm-2。
温度:60℃。
电解时间已经过去之后,在减压下除去溶剂。随后添加10ml乙腈和1ml(0.86g,10.00mmol,0.8当量)哌啶。反应溶液在80℃下在回流下加热12小时。反应时间结束之后减压除去溶剂,并通过柱色谱在硅胶上纯化残余物(环己烷/乙酸乙酯2∶1,RF=0.39)。得到0.01g(0.06mmol,10%)棕色的固体。
1H-NMR(400MHz,DMSO):δ(ppm)=5.41(s,4H,NH2),6.60(dd,3J=7.2Hz,4J=1.1Hz,2H,H-2),7.05(dd,3J=8.4Hz,7.2Hz,2H,H-3)7.17-7.24(m,2H,H-4)。
13C-NMR(101MHz,DMSO):δ(ppm)=107.4(C-2),110.0(C-4),123.8(C-4a),124.3(C-3),144.5(C-1)。
GC(硬法,HP-5):tR=11.89min。
对于C10H11N2计算的HRMS:159.0922;测得:159.0920。
MS(EI,70eV):m/z(%):158(100)[M]·+。
- 还没有人留言评论。精彩留言会获得点赞!