用于确定患者特定突变的药物反应的方法与流程
提供了用于鉴定和确定患者特定突变(patient specific mutation)的药物反应的方法。所述方法允许基于药物对已鉴定的患者特定突变的作用来鉴定特定的(个性化的)药物治疗。
发明背景
癌症(恶性肿瘤或恶性赘生物)是一组涉及具有侵入或扩散到身体其他部分的潜能的异常细胞生长的疾病。癌症极其多样化,并且其涉及多种根本的分子机制。因此,取决于根本的分子机制的准确诊断以及致癌突变的鉴定和自分泌及旁分泌作用,被诊断为患有各种癌症的患者的临床治疗方案和预后可极为不同。在癌症患者中,参与控制细胞生长和分化的许多信号传送途径由于这些途径中关键蛋白中的突变而以异常的方式被调控。
癌症的复杂性和异质性要求更灵敏和识别能力更强的鉴定方法,所述鉴定方法能够模拟肿瘤信号传送途径并且鉴定患者特定驱动突变。例如,属于本申请的发明人的国际公布号WO 2014/111936涉及用于鉴定患者特定驱动突变的方法和系统。目前的技术状态是,仅有很少的单个标志物能够用于预测治疗功效和毒性。此外,由于肿瘤内积累的大量突变、有限的已鉴定的驱动突变的库(repertoire)以及非常有限的关于多种突变的相互作用且特别是其活性的深刻理解,用于选择靶向治疗的全基因组测序(下一代测序)的适用性受到限制。
因此,本领域中存在对以下方法和系统的未满足的需求:所述方法和系统允许鉴定患者特定驱动突变的特定药物反应,以用于确定个性化的且优化的药物治疗,所述方法和系统更加有效、更安全并且有成本和时间效益,并且具有针对患者特定驱动突变被专门调整并优化的能力。
发明概述
本发明提供了用于鉴定和/或确定被调整以适应所鉴定的患者特定致癌突变的优化的药物治疗的方法和系统。通过鉴定受试细胞中与致癌突变的功能相关的信号传送途径活性的改变来识别患者特定致癌突变。根据一些实施方案,通过鉴定与致癌突变的功能相关的报告物基因的亚细胞定位的变化来确定信号传送途径活性的改变。在一些实施方案中,从生物样品中(直接或间接)获得特定患者来源的标志物(patient derived marker)(PDM)基因,并在活的受试细胞中测试它们对相应荧光易位报告物(fluorescent translocation reporter)(FTR)基因的亚细胞易位的影响以确定所测试的PDM是否突变。在一些实施方案中,获得特定患者来源的标志物并将其融合至荧光报告物以创建患者来源的报告物(patient derived reporter)(PDR),其中在活的受试细胞中测试PDR的亚细胞易位以确定所测试的PDR是否突变。在一些实施方案中,致癌突变的鉴定允许测试和确定所鉴定的特定突变对多种药物和/或药物组合的反应(或敏感性)。在一些实施方案中,本文公开的方法允许鉴定优化的药物治疗,所述优化的药物治疗是患者特异性的并且是对于与特定患者突变相关的状况最合适的治疗。
本文公开的方法和系统允许确定或测量药物在PDM或PDR的存在下影响(例如,抑制)所鉴定的异常途径的能力。公开的方法允许检测/鉴定药物反应或抗性以及确定和/或调整适应所鉴定的患者特定致癌突变的最佳个性化治疗。
在一些实施方案中,本发明提供了用于鉴定参与癌症的患者特定致癌突变的方法和系统,并且还提供了用于检测或鉴定所鉴定的致癌突变对多种药物或药物组合的药物反应的方法。在一些实施方案中,突变是致癌突变。在一些实施方案中,突变是驱动突变。在一些实施方案中,所述方法和系统提供了确定所测试的药物或药物组合对患者特定细胞途径和/或特定突变的作用的预测平台。在一些实施方案中,所述方法和系统提供了确定特别适合患者的优化治疗的预测平台。在一些实施方案中,本文公开的方法能够鉴定自分泌和旁分泌对细胞和细胞间的信号传送途径的影响。在一些实施方案中,本文公开的方法能够检测和/或预测固有的耐药机制和获得性的耐药机制。
根据一些实施方案,提供了用于鉴定/确定患者特定致癌突变对药物治疗的药物反应/敏感性的方法,所述方法包括鉴定在受试药物或药物组合的存在和/或不存在下报告物标志物基因的亚细胞定位/易位的改变,其中报告物标志物基因的亚细胞定位的改变受致癌突变的影响。在一些实施方案中,从患者的生物样品中(直接或间接,例如,基于测序数据)获得PDM,并对其操作(工程化)以在报告物嵌合基因(荧光易位报告物(FTR),其包括报告物基因部分和靶基因部分的嵌合产物)的存在下在活的受试细胞中表达。然后确定FTR在受试细胞中的亚细胞定位。如果,与FTR在正常条件下(即,在相应的WT PDM的存在下)的亚细胞定位相比和/或与其他已知的参考相比,FTR在所测试的PDM的存在下的亚细胞定位不同,则指示所测试的PDM发生突变。此外,在受试药物或受试药物组合的存在下测试所鉴定的突变,以鉴定所测试的PDM的特定药物反应(对药物治疗的敏感性)。因此,使用本文公开的方法,还在药物的存在下测定所鉴定的致癌突变以测量所述药物影响所述致癌突变的致癌活性的能力。可选择地或另外地,在一些实施方案中,可以通过创建PDR(即连接/附接/融合至报告物基因的PDM)并且追踪其亚细胞定位直接测试PDM,而无需使用FTR。此外,通过确定这类致癌突变,可鉴定患者肿瘤内运行的已活化的信号传送途径。此外,这促进精确并针对性地选择根除肿瘤并避免特定患者的抗性机制所需要的必需的靶向疗法治疗。
根据一些实施方案,有利地提供了一种用于鉴定特定药物反应以确定优化的个性化的癌症疗法的增强的且改善的诊断平台。在一些实施方案中,所述方法包括基于细胞的测定,所述基于细胞的测定能够通过在存活的(活的)细胞中监测致癌突变对FTR的影响来鉴定活化的致癌突变,并且有效地鉴定和确定所鉴定的突变的药物反应以最终确立患者特定的个性化的药物治疗。本文公开的方法事实上能够有利地以高度显著性预测不同靶向疗法药物的抗性和敏感性,并且能够鉴定多种患者突变的药物反应和对药物治疗的敏感性,并且还提供在多种药物对相同靶标的情况下的药物选择。此外,如下文所示例的,由本文公开的方法鉴定的药物反应还与临床观察到的结果一致,为这些药物的效率提供了机制性解释。因此,这些结果示例了公开的方法和系统鉴定多种突变的药物反应以及在多种药物对相同靶标的情况下提供药物选择的能力。
在一些实施方案中,本文公开的方法和系统提供了一种平台,所述平台能够通过在活的受试细胞中监测多种信号传送蛋白(诸如,例如,膜定位和/或细胞内受体及信号传送蛋白)的活化而鉴定患者肿瘤活化的信号传送途径的特征,并且还确定对多种受试药物或其组合的药物反应(对药物治疗的敏感性)。在一些实施方案中,致癌突变的鉴定可以通过检测涉及FTR的细胞内/亚细胞易位事件(即,易位到多个亚细胞位置,诸如质膜、胞质溶胶、核内体、核仁等/在多个亚细胞位置,诸如质膜、胞质溶胶、核内体、核仁等之间易位)和蛋白-蛋白的相互作用进行。根据一些实施方案,本文公开的方法和系统是有利的,因为除了鉴定同一患者的相同生物样品中的多个突变事件(包括尚未鉴定的突变、以及确定这类突变的致癌活性)以外,还提供了确定这类突变的药物反应的能力,这能够最终允许确定用于特定患者的个性化的治疗方案。因此,本文公开的方法和系统除了允许鉴定导致癌症或参与癌症的细胞事件以外,还能够允许确定或者鉴定特别适合所鉴定的细胞事件和个性化地调整以适应特定患者的有效的优化的药物治疗。
根据一些实施方案,鉴定患者特定突变及其涉及的信号传送途径之后,可以将已知抑制/影响所鉴定的途径/突变的靶向疗法药物/剂(或其组合)与受试细胞孵育,并且可以再次测试抑制的/降低的/改变的致癌活性,以鉴定对肿瘤和患者表现出最佳效果的药物/剂。在一些实施方案中,多种药物/剂不必须已知影响所测试的突变。在一些实施方案中,多种药物/剂治疗及其各自的治疗能力可以被添加到肿瘤致癌图谱上,并且提供患者特定的根本的分子肿瘤机制、治疗选择及其各自期望的用于治疗特定患者的功效。这可以允许医疗保健提供者(health care provider)决定优化的治疗选择。
根据一些实施方案,提供了检测癌症患者的生物样品中一种或更多种患者特定突变的药物反应或者确定癌症患者的生物样品中一种或更多种患者特定突变对药物治疗的敏感性的方法,所述方法包括以下步骤:
a)从生物样品获得多种mRNA;
b)由多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增cDNA文库的特定cDNA;
d)形成扩增的cDNA的单独的表达构建体,其中所述cDNA可操作地连接至启动子;
e)形成携带来自生物样品的扩增的cDNA的第一组表达构建体,及相应的野生型cDNA的第二组表达构建体的可寻址阵列;
f)对于阵列中的每个位点,加入编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
g)将活的测定细胞在使得DNA构建体和载体转染进入测定细胞的条件下添加至每个位点;以及
h)比较表达来自生物样品的cDNA与表达其相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;
i)在药物的存在下重复步骤g),并且比较在药物的存在和不存在下表达来自生物样品的cDNA和/或表达相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;
其中表达源自癌症患者的生物样品的cDNA和表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定生物样品中作为候选的患者特定驱动突变的cDNA;并且其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示候选的患者特定驱动突变的药物反应。
在一些实施方案中,药物是抗癌药物。在一些实施方案中,药物是受试药物。在一些实施方案中,所述方法包括伴随地或顺序地添加多于一种药物。在一些实施方案中,所述方法包括添加药物组合。在一些实施方案中,所述方法包括添加不同浓度的药物以确定药物反应。
在一些实施方案中,FTR的属性选自荧光蛋白的定位及荧光蛋白的易位。在一些实施方案中,定位包括选自以下的亚细胞定位:胞质溶胶、细胞核、核仁、质膜、内质网膜(ER)、线粒体、高尔基体、溶酶体、过氧化物酶体、内体区室(endosomal compartment)和细胞骨架。在一些实施方案中,FTR的靶基因部分编码选自以下的蛋白:肿瘤抑制因子(tumor suppressor)、细胞骨架蛋白、生长因子受体、G-蛋白偶联受体、细胞粘附蛋白、蛋白激酶、转录因子、衔接体蛋白质(adaptor protein)和交换因子(exchange factor)。在另外的实施方案中,FTR的报告物基因部分编码荧光标志物。在一些实施方案中,荧光标志物可以选自:绿色荧光蛋白(GFP)、mCherry、mApple、DsRed、红色荧光蛋白(RFP)、蓝色荧光蛋白(BFP)、EGFP、CFP、YFP、AmCyan1、ZsGreen1、ZsYellow1、DsRed2、AsRed2、和HcRed1。
在一些实施方案中,生物样品选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液。
在一些实施方案中,第一和/或第二组表达构建体包含双链线性DNA。在其他实施方案中,第一和/或第二组表达构建体的启动子是诱导型启动子。在一些实施方案中,第一和/或第二组表达构建体的启动子是组成型启动子。
在一些实施方案中,所述方法还包括对于阵列中的每个位点,诱导表达构建体和/或表达载体在转染细胞中表达以获得来自样品的第一组cDNA的基因产物和FTR。
在另外的实施方案中,扩增的cDNA的表达构建体还包含IRES和第二报告物基因。
在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥DNA构建体。
在一些实施方案中,FTR的表达载体是环状的表达载体。在另外的实施方案中,表达载体包含组成型或诱导型启动子。
在一些实施方案中,步骤g)在步骤e)和/或f)之前,在这些个例中,测定细胞在添加表达构建体和/或表达载体之前被添加至每个位点。
在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥DNA构建体。
根据一些实施方案,提供了鉴定能够抑制一种或更多种患者特定突变的作用的药物的方法,所述方法包括以下步骤:
a)形成携带包含一种或更多种患者特定突变的基因的第一组表达构建体及相应的野生型基因的第二组表达构建体的可寻址阵列;
b)对于可寻址阵列中的每个位点,加入编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至每个位点;以及
d)比较在药物的存在和不存在下表达包含所述一种或更多种患者特定突变的基因与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;
其中在药物的存在和不存在下表达包含一种或更多种患者特定突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示能够抑制一种或更多种患者特定突变的作用的药物。
根据一些实施方案,药物是抗癌药物。在一些实施方案中,所述方法包括伴随地或顺序地添加多于一种药物。在一些实施方案中,所述方法包括添加药物组合。在一些实施方案中,所述方法包括添加不同浓度的药物以确定药物反应。
在一些实施方案中,FTR的属性可以选自荧光蛋白的定位及荧光蛋白的易位。在一些实施方案中,定位可以包括选自以下的亚细胞定位:胞质溶胶、细胞核、核仁、质膜、内质网(ER)、线粒体、高尔基体、溶酶体、过氧化物酶体、内体区室和细胞骨架。
在一些实施方案中,FTR的靶基因部分编码选自以下的蛋白:肿瘤抑制因子、细胞骨架蛋白、生长因子受体、G-蛋白偶联受体、细胞粘附蛋白、蛋白激酶、转录因子、衔接体蛋白质和交换因子。在一些实施方案中,FTR的报告物基因部分可以编码荧光标志物,诸如荧光蛋白。
在一些实施方案中,患者基因可以获自/源自患者的生物样品。在一些实施方案中,生物样品可以选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液、微环境提取物、细胞外液、来自肿瘤的分泌液。在一些实施方案中,患者是癌症患者。在一些实施方案中,突变是致癌突变。
在一些实施方案中,第一和/或第二组表达构建体携带基因的一部分。在一些实施方案中,第一和/或第二组表达构建体包含双链线性DNA。在一些实施方案中,第一和/或第二组表达构建体的启动子可以是诱导型或组成型启动子。在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥表达构建体。在一些实施方案中,步骤c)可在步骤a)和/或b)之前,在这些个例中,测定细胞在添加表达构建体和/或表达载体之前被添加至可寻址阵列。
根据一些实施方案,提供了鉴定一种或更多种患者特定突变对药物治疗的敏感性的方法,所述方法包括以下步骤:a)形成携带包含一种或更多种患者特定突变的基因的第一组表达构建体及相应的野生型基因的第二组表达构建体的可寻址阵列;b)对于可寻址阵列中的每个位点,添加编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;c)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至每个位点;以及d)比较在药物的存在和不存在下表达包含一种或更多种患者特定突变的基因与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;其中在存在和/或不存在药物的情况下表达包含一种或更多种患者特定突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示患者特定突变对用药物治疗的敏感性。
根据一些实施方案,提供了检测癌症患者的一种或更多种患者特定致癌突变的药物反应的方法,所述方法包括以下步骤:
a)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至可寻址阵列中的基底;
b)对于阵列中的每个位点,将荧光易位报告物(FTR)基因的表达载体添加至测定细胞,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)在可寻址阵列的特定位点,将携带包含患者特定突变的基因的第一组表达构建体添加至测定细胞,并且在特定位点,将相应的野生型基因的第二组表达构建体添加至测定细胞,其中第一组表达构建体和第二组表达构建体未被添加至共同位点(common locus);以及
d)比较在药物的存在和不存在下在表达突变与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;
其中在药物的存在和不存在下表达携带突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示患者特定致癌突变的药物反应。
根据一些实施方案,药物是抗癌药物。在一些实施方案中,所述方法包括伴随地或顺序地添加多于一种药物。在一些实施方案中,所述方法包括添加药物组合。在一些实施方案中,所述方法包括添加不同浓度的药物以确定药物反应。
在一些实施方案中,FTR的属性可以选自荧光蛋白的定位及荧光蛋白的易位。在一些实施方案中,定位可以包括选自以下的亚细胞定位:胞质溶胶、细胞核、核仁、质膜、内质网(ER)、线粒体、高尔基体、溶酶体、过氧化物酶体、内体区室和细胞骨架。
在一些实施方案中,FTR的靶基因部分编码选自以下的蛋白:肿瘤抑制因子、细胞骨架蛋白、生长因子受体、G-蛋白偶联受体、细胞粘附蛋白、蛋白激酶、转录因子、衔接体蛋白质和交换因子。在一些实施方案中,FTR的报告物基因部分编码荧光标志物,诸如荧光蛋白。
在一些实施方案中,患者基因可以获自/源自患者的生物样品。在一些实施方案中,生物样品可以选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液、微环境提取物、细胞外液、来自肿瘤的分泌液。在一些实施方案中,患者是癌症患者。在一些实施方案中,突变是致癌突变。
在一些实施方案中,第一和/或第二组表达构建体携带基因的一部分。在一些实施方案中,第一和/或第二组表达构建体包含双链线性DNA。在一些实施方案中,第一和/或第二组表达构建体的启动子可以是诱导型或组成型启动子。在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥表达构建体。在一些实施方案中,步骤b)和/或步骤c)可以在步骤a)之前。
根据一些实施方案,表达构建体可以通过包括以下步骤的一个或更多个的方法获得:i)由获自患者的生物样品的多种mRNA产生cDNA文库;ii)使用与编码疑似携带致癌突变的基因的多核苷酸互补的一组引物扩增cDNA文库的特定cDNA;和iii)将扩增的cDNA可操作地连接至启动子。
在一些实施方案中,可以通过本领域已知的方法合成疑似携带一种或更多种突变的患者基因或基因部分,并且任选地将其可操作地连接至启动子以获得表达构建体。在一些实施方案中,患者基因(或其部分)和/或相应的野生型基因可以基于其序列被人工合成并且被进一步处理以产生相应的PDM。
根据一些实施方案,还提供了用于鉴定患者特定突变及其药物反应的试剂盒。
附图简述
图1是根据一些实施方案的用于鉴定患者驱动突变并检测其药物反应的方法的步骤的示意性框图;
图2是显示根据一些实施方案的鉴定特定患者驱动突变并检测其药物反应的示意图(未按比例);
图3A-显示基于细胞的测定的结果的条形图,其中编码野生型的BRAF(BRAF-WT)或患者突变体BRAF(BRAF突变体,(I554T))的基因已经与报告物蛋白(FTR,ERK2-GFP)一起在受试细胞中表达。细胞未处理或者用ERBB2抑制剂来那替尼(Neratinib)处理。(30小时之后)将细胞固定并且成像,并且使用自动图像分析计算多种实验条件下的核质比(N:C)。
图3B-图3A中示出的所鉴定的突变参与的信号传送途径以及受试药物(来那替尼)的抑制位点的示意图。
图4A-显示基于细胞的测定的结果的条形图,其中编码野生型的BRAF(BRAF-WT)或患者突变体BRAF(BRAF突变体,(I554T))的基因已经与报告物蛋白(FTR,ERK2-GFP)一起在受试细胞中表达。细胞未处理或者用BRAF抑制剂瑞戈非尼(Regorafenib)处理。(30小时之后)将细胞固定并且成像,并且使用自动图像分析计算多种实验条件下的核质比(N:C)。
图4B-图4A中示出的所鉴定的突变参与的信号传送途径以及受试药物(瑞戈非尼)的抑制位点的示意图。
图5A-显示基于细胞的测定的结果的条形图,其中编码野生型形式的EGFR(EGFR-WT)或三突变体形式的EGFR(EGFR三突变体,G719A、T790M和L861Q)的基因已经与报告物蛋白(FTR,ERK2-GFP)一起在受试细胞中表达。细胞未处理或者用EGFR抑制剂厄洛替尼(Erlotinib)处理。(30小时之后)将细胞固定并且成像,并且使用自动图像分析计算多种实验条件下的核质比(N:C)。
图5B-显示基于细胞的测定的结果的条形图,其中编码野生型形式的EGFR(EGFR-WT)或三突变体形式的EGFR(EGFR三突变体,G719A、T790M和L861Q)的基因已经与报告物蛋白(FTR,ERK2-GFP)一起在受试细胞中表达。细胞未处理或者用EGFR抑制剂阿法替尼(Afatinib)处理。(30小时之后)将细胞固定并且成像,并且使用自动图像分析计算多种实验条件下的核质比(N:C)。
图6A-受PDM(BRAF)和相应的FTR(ERK2)影响的信号传送途径以及所测试的药物(维罗非尼(Vemurafenib)、瑞戈非尼和司美替尼(Selumetinib))的抑制位点的示意图。
图6B-显示基于细胞的测定的结果的图,其中编码突变体形式的BRAF(BRAF V600K、BRAF V600E、BRAF G469V)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用BRAF抑制剂维罗非尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图6C-显示基于细胞的测定的结果的图,其中编码突变体形式的BRAF(BRAF V600K、BRAF V600E、BRAF G469V)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用BRAF抑制剂瑞戈非尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图6D-显示基于细胞的测定的结果的图,其中编码突变体形式的BRAF(BRAF V600K、BRAF V600E、BRAF G469V)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用MEK1/2抑制剂司美替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图6E-显示基于细胞的测定的结果的条形图,其中编码突变体形式的BRAF(BRAF V600K、BRAF V600E、BRAF G469V)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用BRAF抑制剂维罗非尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图6F-显示基于细胞的测定的结果的条形图,其中编码突变体形式的BRAF(BRAF V600K、BRAF V600E、BRAF G469V)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用BRAF抑制剂维罗非尼或瑞戈非尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图7A-受PDM(EGFR)和相应的FTR(ERK2、STAT3)影响的信号传送途径以及受试药物(阿法替尼、司美替尼、厄洛替尼、瑞戈非尼)的抑制位点的示意图。
图7B-显示基于细胞的测定的结果的图,其中编码突变体形式的EGFR(EGFR L858R、EGFR L858R/T790M)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用EGFR抑制剂阿法替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图7C-显示基于细胞的测定的结果的图,其中编码突变体形式的EGFR(EGFR L858R、EGFR L861Q)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用MEK1/2抑制剂司美替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图7D-显示基于细胞的测定的结果的图,其中编码突变体形式的EGFR(EGFR L858R、EGFR L861Q、EGFR T790M)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用MEK1/2抑制剂司美替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图7E-显示基于细胞的测定的结果的条形图,其中编码突变体形式的EGFR(EGFR L858R、EGFR G719S/T790M/L861Q)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用EGFR抑制剂阿法替尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图7F-显示基于细胞的测定的结果的条形图,其中编码突变体形式的EGFR(EGFR T790M、L858R、EGFR G719S/T790M/L861Q)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用EGFR抑制剂阿法替尼、BRAF抑制剂瑞戈非尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图8A-受PDM(ERBB2)和相应的FTR(ERK2、STAT3)影响的信号传送途径以及受试药物(拉帕替尼(Lapatinib)、来那替尼、司美替尼)的抑制位点的示意图。
图8B-显示基于细胞的测定的结果的图,其中编码突变体形式的ERBB2(ERBB2V777L、ERBB2S310F)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用ERBB2抑制剂来那替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图8C-显示基于细胞的测定的结果的图,其中编码突变体形式的ERBB2(ERBB2V777L、ERBB2S310F)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用ERBB2抑制剂拉帕替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图8D-显示基于细胞的测定的结果的图,其中编码突变体形式的ERBB2(ERBB2V842I、ERBB2S310F)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用MEK1/2抑制剂司美替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图8E-显示基于细胞的测定的结果的条形图,其中编码突变体形式的ERBB2(ERBB2 V842I、ERBB2 V777L、ERBB2 S310F)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用ERBB2抑制剂来那替尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图8F-显示基于细胞的测定的结果的条形图,其中编码突变体形式的ERBB2(ERBB2 V842I、ERBB2 S310F)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用ERBB2抑制剂来那替尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图9A-受PDM(PIK3CA)和相应的FTR(P38、REL-A)影响的信号传送途径以及受试药物(艾代拉利司(Idelalisib)、Buparlisib、依维莫司(Everolimus)、替西罗莫司(Temsirolimus))的抑制位点的示意图。
图9B-显示基于细胞的测定的结果的图,其中编码突变体形式的PIK3CA(PIK3CA V344A、PIK3CA Q546L、PIK3CA N345K)的基因已经与报告物蛋白(FTR REL-A-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用PIK3CA抑制剂艾代拉利司处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图9C-显示基于细胞的测定的结果的图,其中编码突变形式的PIK3CA(PIK3CA V344A、PIK3CA H1047R、PIK3CA N345K)的基因已经与报告物蛋白(FTR P38-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用PIK3CA抑制剂依维莫司处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图9D-显示基于细胞的测定的结果的图,其中编码突变体形式的PIK3CA(PIK3CA N345K、PIK3CA H1047R)的基因已经与报告物蛋白(FTR P38-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用PIK3CA抑制剂Buparlisib处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图9E-显示基于细胞的测定的结果的条形图,其中编码突变体形式的PIK3CA(PIK3CA N345K、PIK3CA H1047R)的基因已经与报告物蛋白(FTR P38-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用PIK3CA抑制剂替西罗莫司处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图9F-显示基于细胞的测定的结果的条形图,其中编码突变体形式的PIK3CA(PIK3CA N344A、PIK3CA N345K、PIK3CA H1047R)的基因已经与报告物蛋白(FTR P38-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用PIK3CA抑制剂艾代拉利司、依维莫司或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图10A-受PDM(cKIT)和相应的FTR(STAT3)影响的信号传送途径以及受试药物(伊马替尼(Imatinib)和司美替尼)的抑制位点的示意图。
图10B-显示基于细胞的测定的结果的图,其中编码突变体形式的cKIT(cKIT W557-K558del)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用cKIT抑制剂伊马替尼处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图10C-显示基于细胞的测定的结果的图,其中编码突变体形式的cKIT(cKIT W557-K558del)的基因已经与报告物蛋白(FTR STAT3-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用cKIT抑制剂伊马替尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图11A-受PDM(ERBB2和BRAF)和相应的FTR(ERK2)影响的信号传送途径以及所测试的药物(来那替尼、维罗非尼)的抑制位点的示意图。
图11B-显示基于细胞的测定的结果的条形图,其中编码突变体形式的ERBB2(ERBB2 V777L、ERBB2 L755S)或突变体形式的BRAF(BRAF V600E、BRAF V600K)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用ERBB2抑制剂来那替尼、BRAF抑制剂维罗非尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
图12A-受PDM(EGFR和KRAS)和相应的FTR(ERK2)影响的信号传送途径以及受试药物(阿法替尼、司美替尼)的抑制位点的示意图。
图12B-显示基于细胞的测定的结果的条形图,其中编码突变体形式的EGFR(EGFR L858R、EGFR T790M)或突变体形式的KRAS(KRAS G12R)的基因已经与报告物蛋白(FTR ERK2-GFP)一起在受试细胞中表达。细胞未处理(UT)或者用EGFR抑制剂阿法替尼、MEK1/2抑制剂司美替尼或两种抑制剂的组合处理。(24小时之后)将细胞固定并且成像,并且测量细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。
发明详述
根据一些实施方案,提供了用于通过鉴定与驱动突变的功能相关的信号传送途径活性的改变来鉴定或检测患者特定致癌突变的药物反应(对治疗的敏感性)的方法。在一些实施方案中,在受试药物(或者药物组合)的存在和/或不存在下信号传送途径活性的改变通过鉴定报告物基因的亚细胞定位的改变来确定,其中报告物基因的亚细胞定位的改变受驱动突变的影响。在一些实施方案中,患者来源的标志物(PDM)从患者的生物样品中获得,并被操作(工程化)以在报告物嵌合基因(FTR)的存在下在受试细胞中表达。在一些实施方案中,患者来源的标志物(PDM)可以通过基于其所鉴定的序列人工产生(合成)相应的患者基因来获得,并且进一步操作所述患者来源的标志物(PDM)以在受试细胞中表达。在一些实施方案中,另外地或可选择地,将患者特定标志物融合至荧光报告物以创建患者来源的报告物(PDR)。然后确定FTR(和/或PDR,如果可适用)在受试细胞中的亚细胞定位。如果,与FTR(和/或PDR,如果可适用)在正常条件下(即,在相应的WT PDM的存在下)的亚细胞定位相比或与其他预定的参考相比,FTR在所测试的PDM(和/或PDR,如果可适用)的存在下的亚细胞定位不同,则指示所测试的PDM(或PDR)发生突变。此外,如果在受试药物(或药物组合)的存在下进行测定,如果所鉴定的突变的PDM(和/或PDR,如果可适用)的亚细胞定位不同,则所述方法有利地允许精确鉴定或检测所鉴定的PDM(和/或PDR,如果可适用)的药物反应。因此,使用本文公开的方法,可将患者特定PDM鉴定/表征为是致癌突变,并且此外,可确定其对多种受试药物和/或这类受试药物的组合的精确且准确的药物反应(敏感性)。此外,通过确定这类致癌突变,可鉴定患者肿瘤内运行的已活化的信号传送途径及其药物反应。这促进精确并针对性地选择根除肿瘤并避免特定患者的抗性机制所需要的必需的靶向疗法治疗。
在一些实施方案中,本发明基于这样的见解:参与癌症信号传送途径的蛋白响应于多种因子而易位,从而,通过测试在受试药物的存在和/或不存在下受这类信号传送途径影响的嵌合报告物基因的定位,可鉴定患者特定致癌突变并且可鉴定其药物反应。根据一些实施方案,本文公开的方法和系统是有利的,因为,虽然存在有关致癌突变的大量信息,但是之前并不可得用于鉴定同一患者的相同生物样品中的多个突变事件,以及尚未被鉴定的突变,并确定其药物反应的稳健方法和系统。例如,在目前使用的治疗方法中,用格列卫治疗携带cKit突变的胃肠道间质瘤患者。然而,常见的抗性机制通过cKit本身或下游途径中的继发突变(secondary mutation)发生,致使这类治疗无效。同样地,具有EGFR致癌突变的肺癌患者适合于靶向疗法治疗,但是存在若干可用的这类药物。因此,本文公开的方法允许检测和鉴定所鉴定的突变对多种受试药物的反应,以在考虑涉及的特定突变时,最终确定对特定患者的个性化的且优化的药物治疗。
在一些实施方案中,本文公开的方法和系统能够效仿患者肿瘤,以鉴定出已活化的信号传送途径和致癌活性,并且此外确定肿瘤对抗癌疗法的敏感性。在一些实施方案中,这通过将所转染的受试细胞与一种或更多种受试药物孵育来进行。
如本文提及的,术语“多核苷酸分子”、“寡核苷酸”、“多核苷酸”、“核酸”和“核苷酸”序列可以可互换使用。这些术语涉及脱氧核糖核苷酸(DNA)、核糖核苷酸(RNA)及其修饰的形式的聚合物,所述聚合物呈单独的片段,或作为更大的构建体的组分、线性或支链、单链、双链、三链或其杂合体的形式。该术语还包括RNA/DNA杂合体。多核苷酸可包括DNA或RNA的有义和反义寡核苷酸或多核苷酸序列。DNA或RNA分子可以是,例如,但不限于:互补的DNA(cDNA)、基因组DNA、合成的DNA、重组DNA、或其杂合体,或者RNA分子,诸如,例如,mRNA、shRNA、siRNA、miRNA及类似物。因此,如本文使用的,术语“多核苷酸分子”、“寡核苷酸”、“多核苷酸”、“核酸”和“核苷酸”序列意指DNA和RNA分子两者。该术语还包括由天然存在的碱基、糖和共价的核苷间键组成的寡核苷酸,以及具有非天然存在的部分的寡核苷酸,所述非天然存在的部分的功能类似于相应的天然存在的部分。
如本文使用的,术语“构建体”指人工组装或分离的核酸分子,其可包括一个或更多个核酸序列,其中所述核酸序列可包括编码序列(即,编码终产物的序列)、调控序列、非编码序列或其任何组合。术语构建体包括,例如,载体,但不应被视为限于此。
术语“表达载体”指,具有将异源核酸片段(诸如DNA)整合到靶细胞中并使其在靶细胞中表达的能力的载体。换言之,表达载体包含能够被转录的核酸序列/片段。许多病毒、原核和真核表达载体是已知的和/或商购可得的。选择适当的表达载体在本领域技术人员的知识范围内。
如本文使用的,术语“上游”和“下游”指在核苷酸序列,诸如,例如,DNA序列或RNA序列中的相对位置。如众所周知的,核苷酸序列具有5'末端和3'末端,这样的命名是针对核苷酸骨架的糖(脱氧核糖或核糖)环上的碳的。因此,相对于核苷酸序列上的位置,术语下游涉及朝向序列的3'末端的区域。术语上游涉及朝向链的5'末端的区域。
如本文使用的,术语“启动子元件”、“启动子”或“启动子序列”指,通常位于编码序列的5'末端(即,在前面,位于上游)并用作开关活化编码序列的表达的核苷酸序列。如果编码序列被活化,其被表述为被转录。转录通常涉及由编码序列合成RNA分子(诸如,例如,mRNA)。因此,启动子用作转录调控元件并且还提供将编码序列转录为mRNA的起始位点。启动子可完全源自天然来源,或包括源自自然界中发现的不同启动子的不同元件,或甚至包括合成的核苷酸区段。本领域技术人员应理解,不同的启动子可驱动基因在不同的组织或细胞类型中,或在发育的不同阶段或响应不同的环境条件,或以各种表达水平表达。引起基因在大多数细胞类型中在大多数时间表达的启动子通常被称为“组成型启动子”。引起特定组织中的基因表达的启动子被称为“组织特异性启动子”。
如本文使用,术语“引入”和“转染”可互换使用,并指将分子,诸如,例如,核酸、多核苷酸分子、载体和类似物,转移或引入到靶细胞中,且更具体地进入靶细胞的膜包围的空间内部,诸如细胞的胞质溶胶、细胞的细胞核、线粒体的内部空间、内质网(ER)和类似物。分子可以通过本领域技术人员已知的任何方式被“引入”靶细胞中,所述方式例如,如Sambrook等Molecular Cloning:A Laboratory Manual,Cold Spring Harbor Laboratory Press,New York(2001)所教导的,其内容通过引用并入本文。将分子“引入”细胞的方法包括,例如,但不限于:热激、磷酸钙转染、PEI转染、电穿孔、脂质体转染、转染试剂、病毒介导的转移等或其组合。在一些实施方案中,引入的核酸可以是,例如,可呈DNA、RNA形式的修饰的核酸。在一些实施方案中,核酸在被转染至细胞之前被脱水。在一些实施方案中,核酸被整合到载体,诸如,例如,表达载体中。每种可能性代表了本发明的独立的实施方案。
如本文使用,术语“表达”指,期望的终产物分子在靶细胞中的产生。终产物分子可包括例如,RNA分子;肽或蛋白;及类似物;或其组合。
如本文提及的,术语“患者”指具有、疑似具有或诊断为患有疾病的受试者。在一些实施方案中,术语患者指具有、疑似具有或诊断为患有癌症的受试者。在一些实施方案中,患者符合肿瘤活检的条件。
如本文提及的,术语“生物样品”指任何合适的身体来源的样品。样品可包括流体样品诸如全血、外周血单核细胞、白细胞、骨髓。样品可包括各种细胞和组织。样品可包括活检物。样品可包括固定的和/或包埋的组织切片。样品可以是新鲜提取的或冷冻的。在另一个实施方案中,样品是血液样品。在另一个实施方案中,样品是骨髓样品。在另一个实施方案中,用于分离和维持包含来自受试者的血细胞的样品的方法是本领域普通技术人员已知的。在一些实施方案中,包括多核苷酸、多肽、肽、抗体片段及其衍生物的样品可包括体液;细胞制剂的可溶性部分,或细胞生长于其中的培养基;从细胞中分离或提取的染色体、细胞器或膜;溶解的或与基底结合的基因组DNA、RNA或cDNA、多肽、或肽;细胞;组织;组织印记物(tissue print);指纹、皮肤或毛发;其片段或衍生物。在一些实施方案中,生物样品从肿瘤中获得。
如本文提及的,术语“患者来源的标志物”(“PDM”)和“受试者PDM”指从受试者的生物样品中(直接或间接)分离或获得(obtained)或得到(derived)的基因或基因产物(或其部分),并且在功能测定中确认其活性。在一些实施方案中,向PDM核酸序列(其从生物样品直接获得(例如,通过生成cDNA)或被人工合成(即,间接获得))添加5'和/或3'调控元件和/或另外的报告物基因。在一些实例中,如本文中使用的PDM包括嵌合核酸序列分子,所述嵌合核酸序列分子包含调控元件(启动子)-PDM序列-调控元件(IRES)-报告物基因,不必须以该顺序。因此,当这样的核酸分子被引入并表达于靶细胞中时,PDM基因产物(蛋白)和报告物基因产物(蛋白)在该细胞中表达。另外或可选择地,IRES序列可省略,而包含PDM基因产物和报告物基因产物的嵌合蛋白在细胞中表达。由此形成的嵌合蛋白在本文中称为“患者来源的报告物”(“PDR”)或“受试者PDR”。在一些实施方案中,术语“对照PDM”、“野生型PDM”、“相应的PDM”和“相应的野生型PDM”指用作对照的相应于PDM基因的野生型基因(即非突变的、全活性的)。在一些实施方案中,野生型PDM不源自患者的生物样品。使用对照PDM比较受试者PDM与野生型(wt)PDM的活性。
如本文提及的,术语“荧光易位报告物”(“FTR”)指嵌合报告物基因及相应的基因产物。嵌合FTR包含连接至预定靶(标志物)基因部分(诸如,例如,细胞信号传送蛋白、激酶、酶等)的报告物基因部分(诸如荧光标志物(蛋白)),据此靶(标志物)基因的至少一个属性可(直接或间接地)受所测试的PDM影响。
如本文提及的,术语“受试细胞”、“靶细胞”和“测定细胞”可互换使用。这些术语指,被转染了多核酸(poly nucleic acid)分子诸如本文所描述的PDM和/或PDR和/或FTR和/或任何对照基因的测定细胞。在一些实施方案中,受试细胞是真核细胞。在一些实施方案中,受试细胞可以是原代细胞或细胞系。在另一个实施方案中,测定细胞是非癌性细胞。在另一个实施方案中,测定细胞源自细胞系。在另一个实施方案中,测定细胞响应于至少一种癌症分泌的生长因子。在另一个实施方案中,测定细胞适合用转染。在另一个实施方案中,测定细胞适合用瞬时转染。在另一个实施方案中,测定细胞是一个或更多个内源基因的表达已通过任何分子方法被减少或消除的细胞。在另一个实施方案中,测定细胞是Hela细胞。在另一个实施方案中,测定细胞是HEK 293细胞。在另一个实施方案中,测定细胞是PC12细胞。在另一个实施方案中,测定细胞是U2OS细胞。在另一个实施方案中,测定细胞是NCI60细胞系,诸如,A549、EKVX、T47D、HT29。在一些实施方案中,测定细胞是源自患者的细胞。在一些实施方案中,测定细胞是源自癌症患者的细胞。
如本文使用的,术语“亚细胞定位”、“亚细胞区域”和“亚细胞区室”指,细胞的可通过各种方法(诸如,例如,通过可视化方法)与细胞的其他区域区分开的任何限定部分。在一些实例中,亚细胞区域可以是细胞内受限制的区域。在一些实施方案中,亚细胞区域可包括细胞器。亚细胞定位的非限制性实例包括,例如,但不限于:细胞核、核仁、胞质溶胶、线粒体、内质网(ER)、叶绿体、膜、树突棘、高尔基体、溶酶体、过氧化物酶体,内体区室、细胞骨架等。每种可能性是独立的实施方案。在一些实施方案中,术语“亚细胞易位”指,在各种条件下检测到的报告物基因(诸如,FTR或PDR)的亚细胞定位的改变。例如,易位可以是从胞质溶胶至细胞核/从细胞核至胞质溶胶。
如本文提及的,术语“药物”和“受试药物”可互换使用。术语药物指在治疗状况中有效果的任何化合物、物质(substance)、分子、剂和/或试剂。在一些实施方案中,药物是抗癌药物。在一些实施方案中,术语药物可以包括多于一种药物。在一些实施方案中,术语药物包括药物组合。在一些实施方案中,药物是细胞蛋白的抑制剂。
如本文提及的,术语“药物反应”和“对药物的敏感性”可互换使用。该术语指由受试药物引起的反应或效果。在一些实施方案中,该术语涉及药物抑制突变诸如致癌突变的作用的能力。
在一些实施方案中,术语“治疗疾病”或“治疗状况”指,施用一种或更多种有效改善与疾病相关的症状、有效减轻严重程度或治愈该疾病,或有效预防疾病发生的化合物。
术语“检测”、“诊断”指检测疾病、症状、紊乱、病理学或正常状况的方法;疾病、症状、紊乱、病理学状况的分类方法;确定疾病、症状、紊乱、病理学状况的严重程度的方法;监测疾病、症状、紊乱、病理学状况进展的方法;预测其恢复的结果和/或前景的方法。术语“诊断”意指鉴定病理学状况的存在或性质。
术语“基底”指,在其上放置核酸分子、构建体、载体和/或测定细胞的固体支持体。基底可包括任何类型的合适的基底,诸如,但不限于:芯片、载玻片、孔、容器、管、小瓶等。在一些实施方案中,基底是芯片。在一些实施方案中,基底是显微镜载玻片。在一些实施方案中,基底是多孔板,诸如6孔板、12孔板、24孔板、48孔板、96孔板、384孔板等。在一些实施方案中,基底被构造成使得其包括矩阵阵列(位点),由此每个位点(或阵列中的点)被指定并且是可识别的。在一些实施方案中,核酸分子在基底上被脱水。在一些实施方案中,核酸分子在存在或不存在转染试剂时在基底上被脱水。
术语“驱动突变”和“致癌突变”可互换使用。该术语涉及与疾病直接或间接相关的突变的基因或基因产物。在一些实施方案中,该术语涉及与疾病相关和/或参与疾病和/或能够导致和/或引起疾病的突变的基因或基因产物,所述疾病诸如癌症。
术语“编码蛋白的多核苷酸”指编码相应蛋白或其部分的多核苷酸序列或分子。在一些实施方案中,编码蛋白的多核苷酸包含编码相应蛋白的基因或其部分的核苷酸序列。
术语“可寻址阵列”指包括空间上分开的位点的矩阵,所述位点的位置是可鉴定的和可辨识的。在一些示例性实施方案中,可寻址阵列可包括多孔板,其中每个孔(位点)是空间上可鉴定的。在其他示例性实施方案中,可寻址阵列可包括具有可分开的处于/位于指定阵列中的位点的任何基底。
现参考图1,其图示地展示了根据一些实施方案的用于在患者的生物样品中鉴定患者特定致癌突变及用于鉴定其药物反应的方法中的示例性步骤的框图。如图1中所示,在步骤100处,获得患者的生物样品。所述生物样品可选自,但不限于:血液、血清、活检物、穿刺活检物、支气管肺泡灌洗液、胸膜积液、肿瘤组织、尿液、唾液和肿瘤组织。在一些实施方案中,所述生物样品可以是新鲜的(新鲜的或新鲜冷冻的),即,未被固定的样品(步骤102)。在一些实施方案中,所述生物样品可通过本领域已知的用于固定生物样品的方法来固定(步骤104)。
如图1中所示,可从新鲜的生物样品(步骤102)提取多种组分,各自通过本领域熟知的适当方法提取。例如,如步骤106所示,可提取组织间液(interstitial fluid)(IF)(细胞外液)并保存以备将来使用。此外,可从新鲜的生物样品提取mRNA(步骤108)。然后通过本领域熟知的方法(诸如,通过使用polydT引物)使用所提取/分离的mRNA产生cDNA文库(步骤110)。特定的PDM cDNA从cDNA文库扩增并通过使用对应预定的PDM的期望基因区域(多核苷酸)的适当引物对来创建。所选择的PDM可以是基于相应的WT PDM或不同疾病状态中的突变PDM(例如,致癌基因)的已知功能/活性/作用选择的。然后,在步骤112处,通过将调控型启动子元件添加至PDM cDNA的5'末端,并任选地添加3'IRES和标签,诸如报告物基因、荧光标签等来创建测定PDM。在一些实施方案中,启动子元件可以是组成型启动子或诱导型启动子。在一些实施方案中,PDM cDNA还可包括包含FTR编码部分的另外的表达盒。在一些实施方案中,特定的PDM通过人工合成/产生特定的PDM(基于其鉴定的序列)产生,而无需产生cDNA文库的步骤。
还如图1中所示,在步骤114处,可从固定的生物样品(诸如,福尔马林固定的样品(步骤104)中提取基因组DNA。在步骤116处,提取的DNA可经历特殊的、预定的外显子(已知其在癌症病例中发生突变)的扩增,和随后的连接/融合至包含缺少已被扩增的特定外显子的相应的全长基因的表达构建体以产生被测PDM。
然后,在步骤118中,步骤112和/或步骤116中产生(经由产生cDNA,或人工合成)的每种PDM的核酸分子可被放置/点样在支撑性基底(诸如,载玻片、孔(例如,微孔板的孔)、芯片等)上的指定位点(位置)。将PDM放置在具有编码嵌合报告物(FTR)的核酸分子的混合物中,其中选择FTR以对应于PDM(即,选择的FTR在功能上可(直接或间接地)受到PDM影响)。编码PDM和FTR的核酸分子的混合物还可包含适当的转染试剂以允许这些分子被转染到受试细胞。任选地,PDM+FTR混合物被脱水到基底上。在另一种选择中,PDM和FTR被构建为位于单个核酸分子上,以允许两种蛋白在细胞中独立表达。并行地,准备包括WT PDM和相应的FTR的对照测定。此外,在步骤118中,将足量的所选择的受试细胞连同合适的生长培养基一起添加至基底。可在添加核酸分子之前或之后添加细胞。在一些实施方案中,受试细胞的足够量包括约1-10000个细胞/孔(96多孔板的孔)。在一些实施方案中,受试细胞的足够量包括约1-50000个细胞/孔(24多孔板的孔)。在一些实施方案中,受试细胞的足够量包括约1-100000个细胞/孔(12多孔板的孔)。在一些实施方案中,受试细胞的足够量包括约1-1000个细胞/孔(96多孔板的孔)。在一些实施方案中,受试细胞的足够量包括约1-1000个细胞/孔(384多孔板的孔)。在一些实施方案中,受试细胞选自,但不限于:HeLa细胞、HEK 293细胞、U2OS、PC12、NCI60、A549、EKVX、T47D、HT29等。然后将细胞孵育指定的时间段(例如,在约6-60小时的范围内),以允许FTR的表达和任选地PDM的表达。任选地,在一些实施方案中,在步骤118中,将细胞添加至固体基底(具有合适的生长培养基)持续一段时间(诸如0.5-48小时),然后在允许分子转染进入细胞的条件下将编码PDM和/或FTR的核酸分子加入细胞。
然后,在步骤120处,在预定的时间段(诸如,4-60小时)之后,细胞生长培养基可更换为新鲜培养基。在一些实施方案中,更换的培养基是低血清培养基。然后,在另一孵育期(诸如,在4-16小时的范围内)之后,受控于诱导型启动子的PDM的表达的诱导被启动。诱导型启动子的诱导可被启动,例如,当使用四环素诱导型启动子时,可通过添加四环素来启动,或当使用蜕皮激素诱导型启动子时,可通过添加蜕皮激素来启动,或通过本领域已知的任何其他方法来启动。
任选地,在步骤122处,对于从固定的样品中产生的PDM(步骤116),在预定的时间段(诸如,4-60小时)之后,细胞生长培养基更换为新鲜培养基。在一些实施方案中,更换的培养基是低血清培养基。然后,在另一孵育期(诸如,在4-24小时的范围内)之后,PDM在组成型启动子的控制下表达。
然后,在步骤124处,在允许PDM在受试细胞中表达的另一时间段(诸如,例如,在约4-48小时的范围内)之后,确定FTR的亚细胞定位。确定FTR的亚细胞定位可通过各种方法进行,诸如,使用荧光显微镜成像,使用生物化学方法区分亚细胞区室。在一些示例性实施方案中,细胞被固定,并通过荧光成像确定荧光FTR的定位。对FTR在不同实验条件下的亚细胞定位的分析和比较允许确定关于所测试的PDM是否是有缺陷的(即,发生突变)。例如,在共表达FTR与所测试的PDM的细胞中确定FTR的亚细胞定位(测试测定)。此外,在共表达相同的FTR与WT PDM的细胞中确定该FTR的亚细胞定位(对照测定)。测试测定和对照测定的FTR的亚细胞定位之间的差异指示关于所测试的PDM的功能活性。因此,例如,在步骤126中,如果FTR在测试测定中被鉴定为处于与对照测定中相同的亚细胞定位,则所测试的PDM未发生突变。例如,在步骤128中,如果FTR在测试测定中被鉴定为处于与对照测定中不同的亚细胞定位,则所测试的PDM发生突变,这指示该PDM为致癌突变。然后,在步骤130处,为了确定所测试的PDM的药物反应,在受试药物(一种或更多种药物,或者药物组合)的存在下重复步骤120-124,所述受试药物被添加至细胞并且与细胞孵育。可以将药物以不同的浓度(如通过药物的特性所确定的)添加至细胞持续任何时间段,诸如,例如,10分钟至24小时,然后取决于药物类型和有效条件以在1nM至1mM之间的药物浓度进行细胞固定。关于FTR在药物的存在和不存在下定位的不同结果,指示所测试的PDM的药物反应(对药物治疗的敏感性)。
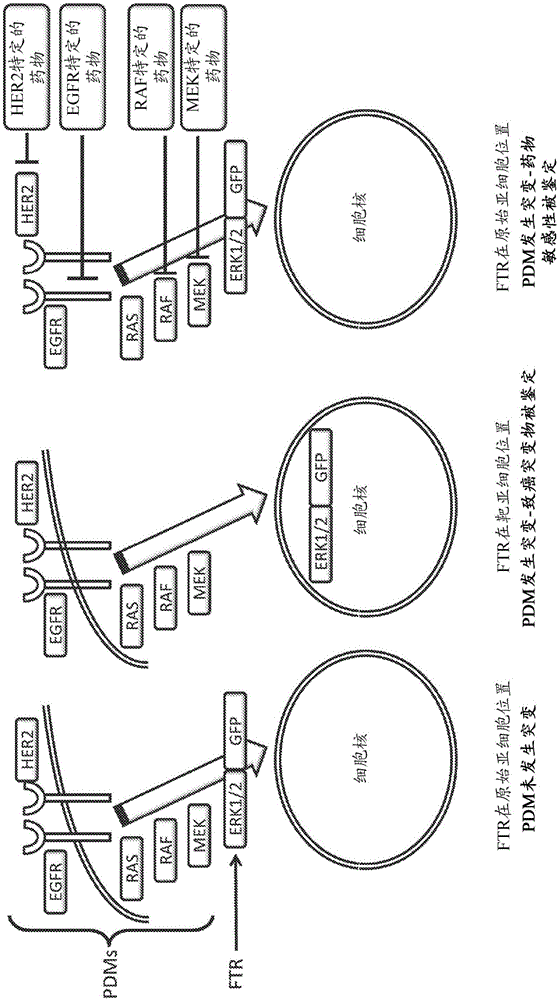
现参考图2,其是根据一些实施方案的应用本发明的方法鉴定示例性细胞信号传送途径中的致癌突变,以及其对受试药物的药物反应的示意图(未按比例)。如图2中所示,为MAP激酶信号传送途径的成员(EGFR、HER2、RAS、RAF、和MEK)的各种PDM按上文描述的,由患者的生物样品制备。该示例性测定中的FTR是包含融合至GFP报告物基因(作为报告物基因部分)的MAPK蛋白(ERK1或ERK2)作为靶(标志物)基因部分的嵌合报告物。PDM和FTR各自如本文以上描述的处理,并确定在不同实验条件下的FTR定位。如左图所示,所测试的PDM无一发生突变,因为检测到的FTR的定位与WT条件中的一样(即,FTR位于细胞质)-因此,所测试的PDM无一发生突变。如中图所示,所测试的PDM中的至少一个发生突变,因为FTR的亚细胞定位与WT条件中不同(即,在该实例中,其在细胞核中)。由于所测试的PDM各自与FTR在不同的受试细胞中被单独地测试,鉴定特定的突变的PDM是可实现的。如右图所示,可以使用影响示例性细胞信号传送途径的多个成员的多种药物,以确定特定药物反应。
根据一些实施方案,提供了用于鉴定癌症患者的生物样品中异常的信号转导途径的药物反应和/或一种或更多种患者特定致癌突变的药物反应的方法,所述方法包括以下步骤的一个或更多个(以任何所选的顺序):
a)从癌症患者的生物样品诸如从肿瘤的活检物获得多种mRNA的样品;
b)通过本领域已知的方法,由多种肿瘤mRNA产生cDNA文库;
c)使用与编码已知蛋白的多核苷酸(基因或基因部分)互补的一组引物扩增所述cDNA文库中的单独的cDNA样品,其中这些蛋白参与多种细胞信号传送途径;
d)形成扩增的cDNA的单独的表达构建体,其中所述cDNA被可操作地连接至启动子以产生受试患者来源的标志物(受试PDM);
e)形成携带来自肿瘤的扩增的cDNA(受试PDM)的第一组表达构建体及平行的相应(相匹配)的野生型蛋白(WT PDM)的第二组表达构建体的可寻址阵列;
f)对于阵列中的每个位点,添加用于共转染与特定报告物基因(FTR)连接的标志物基因的表达载体,其中所述标志物基因受相应的PDM直接或间接影响;
g)任选地在支撑性固体基底上干燥cDNA构建体;
h)将活的测定细胞在使得DNA构建体转染进入测定细胞的条件下添加至每个位点;
i)对于阵列中的每个位点,允许构建体和表达载体在转染的细胞中表达,以获得来自肿瘤的第一组cDNA和特定报告物基因的基因产物;
j)比较表达来自肿瘤的cDNA与表达其相应的野生型表达的cDNA的细胞中的报告物基因的至少一个属性;
k)在药物的存在下重复步骤h)至i)中的任一个,并且比较在药物的存在和不存在下表达来自肿瘤的cDNA和/或表达相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;
其中表达源自癌症患者的生物样品的cDNA和表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定来自生物样品的cDNA作为候选的患者特定致癌突变;并且其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示候选的患者特定致癌突变的药物反应。
在一些实施方案中,PDM的表达盒和FTR的表达盒位于一个表达构建体上(即,在一个分子上)。在这类实施方案中,PDM表达盒和FTR表达盒可具有相同、相似或不同的启动子(即,PDM和FTR的表达可由相同或不同的启动子来控制)。在这类实施方案中,上文的步骤d)和f)被组合成一个步骤:(替代性的步骤d):形成扩增的cDNA的单独的表达构建体,其中cDNA被可操作地连接至启动子,以产生受试患者来源的标志物(受试PDM);其中所述表达构建体还包括特定报告物基因(FTR)。在一些实施方案中,FTR被连接至启动子(其可与PDM的启动子相同或不同)。在另外的实施方案中,FTR包含与报告物基因部分连接的靶基因部分。
在一些实施方案中,步骤g)在步骤e)和/或f)之前,在这些个例中,测定细胞在添加表达盒之前被添加至每个位点。
在一些实施方案中,所述方法在步骤e)中包括相应的PDM蛋白的第三组表达构建体,所述相应的PDM蛋白包含所述基因中的一种或更多种已知的驱动突变(本文中的“人工PDM”),其可以被用作实验对照。将第三组表达构建体添加至可寻址阵列。
因此,根据一些实施方案,提供了鉴定癌症患者的生物样品中的一种或更多种患者特定致癌突变的药物反应的方法,所述方法包括以下步骤的一个或更多个:
a)从生物样品获得多种mRNA;
b)由多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增cDNA文库中的特定cDNA;
d)形成扩增的cDNA的单独的表达构建体,其中cDNA被可操作地连接至启动子;
e)形成携带来自生物样品的扩增的cDNA的第一组表达构建体及相应的野生型cDNA的第二组表达构建体的可寻址阵列;
从而提供了在编码信号转导蛋白的多核苷酸中携带候选突变的表达构建体的可寻址阵列,所述阵列适合于在所述癌症患者的生物样品中鉴定患者特定致癌突变。
在一些实施方案中,所述方法还包括步骤(f):对于阵列中的每个位点,添加编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分。
在另外的实施方案中,所述方法还包括以下步骤:(g)将活的测定细胞在使得DNA构建体和载体转染进入测定细胞的条件下添加至每个位点;以及(h)比较表达来自样品的cDNA与表达其相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;其中,表达源自癌症患者的生物样品的cDNA与表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定来自生物样品的cDNA作为候选的患者特定致癌突变。
在另外的实施方案中,所述方法还包括步骤i),所述步骤i)包括在药物的存在下重复步骤g)至h)中的任一个,并且比较在药物的存在和不存在下表达来自样品的cDNA和/或表达相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;其中表达源自癌症患者的生物样品的cDNA与表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定来自生物样品的cDNA作为候选的患者特定致癌突变;并且其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示候选的患者特定致癌突变的药物反应。
在一些实施方案中,所述方法在步骤e)中包括相应的PDM蛋白的第三组表达构建体,所述相应的PDM蛋白包含所述基因中的一种或更多种已知的驱动突变(本文中的“人工PDM”),其可以被用作实验对照。将第三组表达构建体添加至可寻址阵列。
根据一些实施方案,提供了鉴定或确定癌症患者的生物样品中的一种或更多种患者特定致癌突变的药物反应的方法,包括以下步骤:
a)从生物样品获得多种mRNA;
b)由多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增cDNA文库中的特定cDNA;
d)形成扩增的cDNA的单独的表达构建体,其中cDNA被可操作地连接至启动子;
e)在可寻址阵列中,将活的测定细胞加至基底上;
f)将携带来自生物样品的扩增的cDNA的第一组表达构建体和携带相应的野生型cDNA的第二组表达构建体添加至测定细胞;其中在使得表达构建体转染进入测定细胞的条件下,将每种表达构建体添加至位于不同的可寻址位点处的测定细胞;
从而产生包含在编码信号转导蛋白的多核苷酸中携带候选突变的表达构建体的测定细胞的阵列,所述阵列用于在所述癌症患者的生物样品中鉴定患者特定致癌突变。
在一些实施方案中,所述方法还包括对于阵列中的每个位点,将荧光易位报告物(FTR)基因的表达载体添加至测定细胞的步骤,所述荧光易位报告物(FTR)基因包括连接至特定报告物基因部分的靶基因部分。
在一些实施方案中,用指定的FTR稳定地转染测定细胞,并且可以将这类表达相应的FTR的测定细胞用于本文公开的方法中。
在一些实施方案中,所述方法在步骤f)中包括在使得转染的条件下向位于不同的可寻址位点处的测定细胞添加相应的PDM蛋白的第三组表达构建体,所述相应的PDM蛋白包含所述基因中的一种或更多种已知的驱动突变(相应的人工PDM)。
在一些实施方案中,所述方法还包括,比较表达来自生物样品的cDNA与表达其相应的野生型表达的cDNA和/或表达相应的人工PDM的细胞中的FTR的至少一个属性;其中,表达生物样品来源的cDNA与表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定来自生物样品的cDNA作为候选的患者特定致癌突变。
在一些实施方案中,所述方法还包括重复以下的步骤:将药物添加至测定细胞并且比较在药物的存在和不存在下在表达来自生物样品的cDNA和/或表达相应的野生型表达的cDNA和/或表达相应的人工PDM的测定细胞中表达的FTR的至少一个属性;其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示候选的患者特定致癌突变的药物反应。
在一些实施方案中,生物样品选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液。
根据一些实施方案,提供了用于鉴定癌症患者的生物样品中的异常信号转导途径的药物反应的方法,和/或用于鉴定患者特定致癌突变的药物反应的方法,所述方法包括以下步骤的一个或更多个(以任何适当的顺序):
a)从癌症患者的生物样品,诸如从肿瘤的活检物获得多种mRNA样品;
b)通过本领域已知的方法,由多种肿瘤mRNA产生cDNA文库;
c)使用与编码已知蛋白的多核苷酸互补的一组引物扩增cDNA文库的单独的cDNA样品,其中所述蛋白参与多种细胞信号传送途径;
d)形成扩增的cDNA的单独的表达构建体,其中所述cDNA被可操作地连接至启动子和报告物基因,以产生嵌合的受试患者来源的报告物(受试PDR);
e)形成携带来自肿瘤的扩增的cDNA(受试PDR)的第一组表达构建体及平行的cDNA(wt PDR)的第二组表达构建体的可寻址阵列;
f)任选地在支撑性固体基底上干燥cDNA构建体;
g)将活的测定细胞在使得DNA构建体转染进入测定细胞的条件下添加至每个位点;
h)允许构建体和表达载体在转染的细胞中表达,以获得第一组来自肿瘤的cDNA的基因产物;
i)比较表达来自肿瘤的cDNA与表达其相应的野生型表达的cDNA的细胞中的嵌合报告物基因的至少一个属性;
j)在药物的存在下重复步骤g)至h)中的任一个,并且比较在药物的存在和不存在下表达来自肿瘤的cDNA和/或表达相应的野生型表达的cDNA和/或表达相应的人工PDM的测定细胞中表达的FTR的至少一个属性;
其中表达肿瘤来源的cDNA和表达相应的野生型cDNA的细胞之间的不同结果用于鉴定来自肿瘤的cDNA作为候选的异常信号转导蛋白;并且其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示候选的患者特异致癌突变的药物反应。
根据一些实施方案,提供了检测或鉴定肿瘤细胞中的异常信号转导途径的药物反应的方法,所述方法包括以下步骤的一个或更多个(以任何适当的顺序):
a)从可在体外或体内获得,例如,从肿瘤活检物获得的肿瘤细胞获得mRNA样品;
b)由获得的多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增cDNA文库中的单独的cDNA样品;
d)形成扩增的cDNA的单独的表达构建体,其中cDNA被可操作地连接至启动子;
e)形成携带来自肿瘤的扩增的cDNA的第一组表达构建体及平行的相应的野生型cDNA的第二组表达构建体,以及任选地相应的PDM蛋白的第三组表达构建体的可寻址阵列,所述相应的PDM蛋白包含所述基因中的一种或更多种已知的驱动突变(人工PDM);
f)对于阵列中的每个位点,添加用于共转染荧光易位报告物(FTR)嵌合基因的表达载体,所述荧光易位报告物(FTR)嵌合基因包含连接至报告物基因部分的靶基因部分;
g)将活的测定细胞在使得DNA构建体转染进入测定细胞的条件下添加至每个位点;
h)比较表达来自肿瘤的cDNA与表达其相应的野生型表达的cDNA的细胞中表达的FTR的至少一个属性;
i)在药物的存在下重复步骤g)至h)中的任一个,并且比较在药物的存在和不存在下表达来自肿瘤的cDNA和/或表达相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;
其中表达肿瘤来源的cDNA和表达相应的野生型cDNA的细胞之间的不同结果用于鉴定来自肿瘤的cDNA作为候选的异常信号转导蛋白;并且其中在药物的存在和不存在下表达源自癌症患者的生物样品的cDNA的测定细胞之间的不同结果指示异常信号转导途径的药物反应。
在一些实施方案中,药物是抗癌药物/剂。在一些实施方案中,将多于一种药物添加至细胞。在一些实施方案中,可以将药物组合(混合物)添加至细胞。在一些实施方案中,当将多于一种药物添加至细胞时,可以以任何时间间隔伴随地或顺序地添加药物并且持续任何孵育时间段。
在一些实施方案中,PDM的表达盒和FTR的表达盒位于一个表达构建体上(即,在一个分子上)。在这类实施方案中,PDM表达盒和FTR表达盒可具有相同、相似或不同的启动子(即,PDM和FTR的表达可由相同或不同的启动子来控制)。在一些实施方案中,步骤g)可在步骤e)和/或f)之前,在这些个例中,测定细胞在添加表达构建体和/或表达载体之前被添加至每个位点。
因此,根据一些实施方案,提供了鉴定肿瘤细胞中的异常信号转导途径的药物反应的方法,包括以下步骤的一个或更多个(以任何适当的顺序):
a)从可在体外或体内获得,例如,从肿瘤活检物获得的肿瘤细胞获得mRNA样品;
b)由获得的多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增所述cDNA文库中的单独的cDNA样品;
d)形成扩增的cDNA的单独的表达构建体,其中所述cDNA被可操作地连接至第一启动子;所述表达构建体还包含含有第二启动子并编码荧光易位报告物(FTR)嵌合基因的表达盒,所述FTR包含连接至报告物基因部分的靶基因部分;
e)形成携带来自肿瘤的扩增的cDNA和FTR盒的第一组表达构建体及平行的相应的野生型cDNA和FTR盒的第二组表达构建体的可寻址阵列;
f)将活的测定细胞在使得DNA构建体转染进入测定细胞的条件下添加至每个位点;以及
g)比较表达来自肿瘤的cDNA与表达其相应的野生型表达的cDNA的细胞中表达的FTR的至少一个属性;
其中,表达肿瘤来源的cDNA与表达相应的野生型cDNA的细胞之间的不同结果用于鉴定来自肿瘤细胞的cDNA作为候选的异常信号转导蛋白。
在一些实施方案中,第一和第二启动子是相同或不同的。
根据一些实施方案,提供了鉴定肿瘤细胞中的异常信号转导途径的药物反应的方法,所述方法包括以下步骤:
a)从肿瘤细胞获得多种mRNA;
b)由多种mRNA产生cDNA文库;
c)使用与编码已知的信号转导蛋白的多核苷酸互补的一组引物扩增cDNA文库的特定cDNA;
d)形成步骤(c)中扩增的cDNA的单独的表达构建体,其中所述cDNA被可操作地连接至启动子;
e)形成携带来自肿瘤的扩增的cDNA的第一组表达构建体及相应的野生型cDNA的第二组表达构建体的可寻址阵列;
从而提供在编码信号转导蛋白的多核苷酸中携带候选突变的表达构建体的可寻址阵列,所述可寻址阵列适合于鉴定所述肿瘤细胞中异常的信号转导途径。
在一些实施方案中,所述方法还包括步骤(f):对于阵列中的每个位点,添加编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至报告物基因部分的靶基因部分。
在另外的实施方案中,所述方法还包括以下步骤:g)将活的测定细胞在使得DNA构建体共转染进入测定细胞的条件下添加至每个位点;以及h)比较表达来自肿瘤的cDNA与表达其相应的野生型表达的cDNA的测定细胞中表达的FTR的至少一个属性;其中,表达源自肿瘤细胞的cDNA与表达相应的野生型cDNA的测定细胞之间的不同结果用于鉴定来自肿瘤细胞的cDNA作为候选的异常信号转导蛋白。
根据一些实施方案,提供了用于鉴定癌症患者的生物样品中能够影响致瘤性(tumoriogenity)的因子的药物反应的方法,所述方法包括以下步骤的一个或更多个(以任何适当的顺序):
a)在可寻址阵列中,将活的测定细胞添加至基底;
b)对于阵列中的每个位点,将荧光易位报告物(FTR)基因的表达载体添加至测定细胞,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)在阵列的特定位点将患者的生物样品添加至测定细胞;
d)比较其中添加了患者的生物样品的细胞与其中未添加生物样品的相应细胞中表达的FTR的至少一个属性;
e)在药物的存在下重复步骤c)-d)中的任一个,并且比较在药物的存在和不存在下其中添加了患者的生物样品的测定细胞与其中未添加生物样品的相应细胞中表达的FTR的至少一个属性;
其中将其中添加了患者的生物样品的细胞与其中未添加生物样品的相应细胞之间的不同结果用于鉴定生物样品包含能够影响致瘤性的因子;并且其中在药物的存在和不存在下其中添加了患者的生物样品的细胞和其中未添加生物样品的相应细胞之间的不同结果指示能够影响致瘤性的因子的药物反应。
在一些实施方案中,生物样品选自患者肿瘤微环境、细胞外液、来自肿瘤的分泌液、血浆、支气管肺泡灌洗液及类似物或其组合。
在一些实施方案中,能够影响致瘤性的因子选自自分泌因子、旁分泌因子或两者。
在一些实施方案中,测定细胞稳定表达FTR。在一些实施方案中,步骤b)可以在步骤a)之前,在这些个例中,将细胞添加至已添加在可寻址阵列中的FTR的表达载体。
根据一些实施方案,提供了鉴定能够抑制一种或更多种患者特定突变的作用的药物的方法,和/或鉴定一种或更多种患者特定突变的药物反应的方法,和/或鉴定一种或更多种患者特定突变对药物的敏感性的方法,所述方法包括以下步骤的一个或更多个(以任何顺序):
a)形成携带包含患者特定突变的基因的第一组表达构建体及相应的野生型基因的第二组表达构建体的可寻址阵列;
b)对于可寻址阵列中的每个位点,添加编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至每个位点;以及
d)比较在药物的存在和不存在下表达包含所述一种或更多种患者特定突变的基因与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;
其中在药物的存在和不存在下表达包含突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示能够抑制患者特定突变的作用的药物。
根据一些实施方案,药物是抗癌药物。在一些实施方案中,所述方法包括伴随地或顺序地添加多于一种药物。在一些实施方案中,所述方法包括添加药物组合。在一些实施方案中,所述方法包括添加不同浓度的药物以确定药物反应。
在一些实施方案中,FTR的属性可以选自荧光蛋白的定位及荧光蛋白的易位。在一些实施方案中,定位可以包括选自以下的亚细胞定位:胞质溶胶、细胞核、核仁、质膜、内质网(ER)、线粒体、高尔基体、溶酶体、过氧化物酶体、内体区室和细胞骨架。
在一些实施方案中,FTR的靶基因部分编码选自以下的蛋白:肿瘤抑制因子、细胞骨架蛋白、生长因子受体、G-蛋白偶联受体、细胞粘附蛋白、蛋白激酶、转录因子、衔接体蛋白质和交换因子。在一些实施方案中,FTR的报告物基因部分可以编码荧光标志物,诸如荧光蛋白。
在一些实施方案中,患者基因可以获自/源自患者的生物样品。在一些实施方案中,生物样品可以选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液、微环境提取物、细胞外液、来自肿瘤的分泌液。在一些实施方案中,患者是癌症患者。在一些实施方案中,突变是致癌突变。
在一些实施方案中,第一和/或第二组表达构建体携带基因的一部分。在一些实施方案中,第一和/或第二组表达构建体包含双链线性DNA。在一些实施方案中,第一和/或第二组表达构建体的启动子可以是诱导型或组成型启动子。在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥表达构建体。在一些实施方案中,PDM的表达盒和FTR的表达盒位于一个表达构建体上(即,在一个分子上)。在这类实施方案中,PDM表达盒和FTR表达盒可具有相同、相似或不同的启动子(即,PDM和FTR的表达可由相同或不同的启动子来控制)。在一些实施方案中,步骤c)可在步骤a)和/或b)之前,在这些个例中,测定细胞在添加表达构建体和/或表达载体之前被添加至可寻址阵列。
根据一些实施方案,提供了鉴定一种或更多种患者特定突变对药物治疗的敏感性的方法,所述方法包括以下步骤:
a)形成携带包含患者特定突变的基因的第一组表达构建体及相应的野生型基因的第二组表达构建体的可寻址阵列;
b)对于可寻址阵列中的每个位点,添加编码荧光易位报告物(FTR)基因的表达载体,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至每个位点;以及
d)比较在药物的存在和不存在下在表达包含突变的基因与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;
其中在药物的存在和不存在下表达携带突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示患者特定致癌突变对用药物治疗的敏感性。
根据一些实施方案,提供了检测癌症患者的一种或更多种患者特定致癌突变的药物反应的方法,所述方法包括以下步骤:
a)将活的测定细胞在使得表达构建体和表达载体转染进入测定细胞的条件下添加至基底;
b)对于阵列中的每个位点,将荧光易位报告物(FTR)基因的表达载体添加至测定细胞,所述荧光易位报告物(FTR)基因包含连接至特定报告物基因部分的靶基因部分;
c)在可寻址阵列的特定位点,将携带包含患者特定突变的基因的第一组表达构建体添加至测定细胞,并且在特定位点,将相应的野生型基因的第二组表达构建体添加至测定细胞,其中第一组表达构建体和第二组表达构建体未被加入至共同位点;以及
d)比较在药物的存在和不存在下在表达致癌突变与表达其相应的野生型表达的基因的测定细胞中表达的FTR的至少一个属性;
其中在不存在和/或存在药物的情况下表达携带突变的基因和/或表达相应的野生型基因的测定细胞之间的不同结果指示患者特定驱动突变的药物反应。
根据一些实施方案,药物是抗癌药物。在一些实施方案中,所述方法包括伴随地或顺序地添加多于一种药物。在一些实施方案中,所述方法包括添加药物组合。在一些实施方案中,所述方法包括添加不同浓度的药物以确定药物反应。
在一些实施方案中,FTR的属性可以选自荧光蛋白的定位及荧光蛋白的易位。在一些实施方案中,定位可以包括选自以下的亚细胞定位:胞质溶胶、细胞核、核仁、质膜、内质网(ER)、线粒体、高尔基体、溶酶体、过氧化物酶体、内体区室和细胞骨架。
在一些实施方案中,FTR的靶基因部分编码选自以下的蛋白:肿瘤抑制因子、细胞骨架蛋白、生长因子受体、G-蛋白偶联受体、细胞粘附蛋白、蛋白激酶、转录因子、衔接体蛋白质和交换因子。在一些实施方案中,FTR的报告物基因部分可以编码荧光标志物,诸如荧光蛋白。
在一些实施方案中,患者基因可以获自/源自患者的生物样品。在一些实施方案中,生物样品可以选自肿瘤细胞、肿瘤活检物、肿瘤组织和体液、微环境提取物、细胞外液、来自肿瘤的分泌液。在一些实施方案中,患者是癌症患者。在一些实施方案中,突变是致癌突变。
在一些实施方案中,第一和/或第二组表达构建体携带基因的一部分。在一些实施方案中,第一和/或第二组表达构建体包含双链线性DNA。在一些实施方案中,第一和/或第二组表达构建体的启动子可以是诱导型或组成型启动子。在一些实施方案中,所述方法还包括在转染试剂的存在下在固体支持体上干燥表达构建体。在一些实施方案中,步骤b)和/或步骤c)可以在步骤a)之前,在这些个例中,细胞可以被添加至表达构建体和/或表达载体。
根据一些实施方案,本文公开的方法中使用的表达构建体可以通过包括以下步骤的一个或更多个的方法获得:
i)由获自患者的生物样品的多种mRNA产生cDNA文库;
ii)使用与编码疑似携带致癌突变的基因的多核苷酸互补的一组引物扩增cDNA文库的特定cDNA;以及
iii)将扩增的cDNA可操作地连接至启动子。
在一些实施方案中,可以通过本领域已知的方法合成疑似携带一种或更多种突变的患者的基因或基因部分,并且任选地将其可操作地连接至启动子以获得表达构建体。在一些实施方案中,如本文所详述的,患者基因(或其部分)和/或相应的野生型基因可以基于其序列被人工合成并且被进一步处理以产生相应的PDM。
根据一些实施方案,患者是罹患癌症的患者。在一些实施方案中,癌症包括如下这些癌症:癌(carcinomas)、肉瘤、骨髓瘤、白血病、淋巴瘤和混合型肿瘤。特定类别的肿瘤包括淋巴组织增生性紊乱、乳腺癌、卵巢癌、前列腺癌、宫颈癌、子宫内膜癌、骨癌、肝癌、胃癌、结肠癌、肺癌、胰腺癌、甲状腺癌、头颈癌、中枢神经系统癌、周围神经系统癌、皮肤癌、肾癌,以及上述所有的转移病灶。适合治疗的特定类型的肿瘤包括:肝细胞癌、肝细胞瘤(hepatoma)、肝母细胞瘤、横纹肌肉瘤、食管癌、甲状腺癌、成神经节细胞瘤(ganglioblastoma)、纤维肉瘤、粘液肉瘤、脂肪肉瘤、软骨肉瘤、骨原性肉瘤、脊索瘤、血管肉瘤、内皮肉瘤、尤因氏瘤、平滑肌肉瘤、rhabdotheliosarcoma、浸润性导管癌、乳头状腺癌、黑色素瘤、鳞状细胞癌、基底细胞癌、腺癌(高度分化、中度分化、低度分化或未分化)、肾细胞癌、肾上腺样瘤、肾上腺样腺癌(hypernephroid adenocarcinoma)、胆管癌、绒毛膜癌、精原细胞瘤、胚胎癌、肾母细胞瘤(Wilms’tumor)、睾丸肿瘤、包括小细胞肺癌、非小细胞肺癌和大细胞肺癌的肺癌、膀胱癌、神经胶质瘤、星形细胞瘤、髓母细胞瘤、颅咽管瘤、室管膜瘤、松果体瘤、视网膜母细胞瘤、神经母细胞瘤、结肠癌、直肠癌、包括所有类型的白血病和淋巴瘤的造血系统恶性肿瘤,包括:急性骨髓性白血病、急性髓细胞白血病、急性淋巴细胞白血病、慢性骨髓性白血病、慢性淋巴细胞白血病、肥大细胞白血病、多发性骨髓瘤、髓性淋巴瘤、霍奇金氏淋巴瘤、非霍奇金氏淋巴瘤。
根据某些实施方案,癌症选自前列腺癌、乳腺癌、皮肤癌、结肠癌、肺癌、胰腺癌、淋巴瘤、骨髓瘤、白血病、头颈癌、肾癌、卵巢癌、骨癌、肝癌或甲状腺癌。
在一些实施方案中,患者已被诊断为癌症阳性。在一些实施方案中,患者被施以具有已知或未知的治疗效果的靶向疗法的治疗方案。在一些实施方案中,患者有可利用的患者肿瘤分子学特征谱(IHC、FISH、PCR及测序)。在一些实施方案中,患者有可利用的病历(patient history)以及治疗结果(患者反应、耐受性、复发率和存活率)。
在一些实施方案中,生物样品选自:血液、血清、活检物、组织、穿刺活检物、支气管肺泡灌洗液、胸膜积液、尿液、唾液和肿瘤。在一些实施方案中,生物样品可以是新鲜分离的。在一些实施方案中,生物样品可以是冷冻的。在一些实施方案中,生物样品可被固定。
在一些实施方案中,在测定细胞中表达的每种蛋白(诸如,所测试的PDM、FTR、WT PDM、PDR)是可区别性地鉴定的。在另一个实施方案中,每种蛋白可直接或间接地通过不同的标志物或不同的报告物或不同的荧光蛋白鉴定。在另一个实施方案中,每种嵌合蛋白(诸如,FTR或PDR)包含不同的报告物部分。在另一个实施方案中,不同的蛋白可都有荧光蛋白或报告物。在另一个实施方案中,本发明的每种嵌合蛋白包括不同的报告物部分。
在另一个实施方案中,PDM与癌症的生长相关。在另一个实施方案中,PDM是致癌基因或肿瘤抑制因子。在另一个实施方案中,PDM是细胞骨架调控因子。在另一个实施方案中,PDM在肿瘤生长和转移方面起作用。在另一个实施方案中,PDM是囊泡运输蛋白。在另一个实施方案中,PDM是囊泡拴系蛋白(vesicle tethering protein)。在另一个实施方案中,PDM是细胞粘附蛋白。在另一个实施方案中,PDM是核完整性蛋白(nuclear integrity protein)。在另一个实施方案中,PDM是生长因子受体。在另一个实施方案中,PDM是细胞因子受体。在另一个实施方案中,PDM是细胞粘附蛋白。在另一个实施方案中,PDM参与肿瘤炎症。在另一个实施方案中,PDM是细胞极性蛋白。在另一个实施方案中,PDM是信号传送蛋白。在另一个实施方案中,PDM是衔接体蛋白质。在另一个实施方案中,PDM是蛋白激酶。在另一个实施方案中,PDM是交换因子。在另一个实施方案中,PDM是细胞骨架蛋白。在一些示例性实施方案中,PDM选自包括以下的组或由以下组成的组:AKT1、AKT2、AKT3、ALK、BRAF、BRCA1、BRCA2、CBL、CTNNB1、EGFR、ERBB2、ERBB3、FGFR1、FGFR2、GNA11、GNAQ、HRAS、JAK2、KIT、KRAS、MET、NRAS、PDGFRA、PIK3CA、PTEN、RAF1、RET、ROS1、SMO、TP53、SMAD2、SMAD3、SMAD4、STAT1、STAT3、STAT5B、TGFBR2、FBXW7、MYC、LKB1、SMARCA4、TCF7L2、MAP3K1、ESR1、AR、PR、DDR2、MEK1或其任何组合。每种可能性是独立的实施方案。
在另一个实施方案中,PDM与标志物(标签)诸如荧光蛋白(诸如mCherry、mApple、GFP、Cherry、DsRed、RFP、EGFP、BFP、YFP、AmCyan1、ZsGreen1、ZsYellow1、DsRed2、AsRed2、和HcRed1)联合表达。在一些实施方案中,标志物包含Cys-Cys-Pro-Gly-Cys-Cys(SEQ ID NO:47)的标志物基序,并在成像之前,可向测试性测定添加FlAsH-EDT2或ReAsH-EDT2,以在结合包含Cys-Cys-Pro-Gly-Cys-Cys基序的重组蛋白后发出荧光。在一些实施方案中,包含Cys-Cys-Pro-Gly-Cys-Cys的蛋白可以是PDM、单独的荧光蛋白、或与可进一步用于标记亚细胞性细胞器,诸如,例如,质膜或细胞核的亚细胞标志物融合的荧光蛋白。在一些实施方案中,与PDM联合表达的标志物(标签)用作验证测定细胞中PDM的转染和表达的标志物。
在另一个实施方案中,PDR是与标志物(标签)诸如荧光蛋白(诸如mCherry、mApple、GFP、Cherry、DsRed、RFP、EGFP、BFP、YFP、AmCyan1、ZsGreen1、ZsYellow1、DsRed2、AsRed2、和HcRed1)融合的PDM。在一些实施方案中,PDR是与包含Cys-Cys-Pro-Gly-Cys-Cys(SEQ ID NO:47)基序的标志物(标签)融合的PDM。
在一些实施方案中,FTR是融合(嵌合)蛋白,其包含报告物部分,诸如荧光蛋白(诸如mCherry、mApple、GFP、樱桃红、DsRed、RFP、EGFP、BFP、YFP、AmCyan1、ZsGreen1、ZsYellow1、DsRed2、AsRed2、和HcRed1)或Cys-Cys-Pro-Gly-Cys-Cys基序,和选自但不限于以下的靶蛋白部分:与癌症生长相关的蛋白、致癌基因产物、细胞骨架调控因子、囊泡运输蛋白、囊泡拴系蛋白、细胞粘附蛋白、核完整性蛋白、生长因子受体、细胞粘附蛋白、细胞信号传送蛋白、参与肿瘤炎症的蛋白、细胞极性蛋白、生长因子信号传送蛋白、衔接体蛋白质、细胞骨架蛋白等。每种可能性是独立的实施方案。
在一些示例性实施方案中,FTR是包含报告物部分诸如荧光蛋白和靶(标志物)蛋白部分的融合蛋白,所述靶(标志物)蛋白部分选自但不限于包括以下的组或者由以下组成的组:AKT1、AKT2、mTOR、RelA、NFKB1、NFKB2、ERK1、ERK2、ERF、STAT1、STAT3、STAT5、CTNNB1、JNK1α、JNK1β、JNK2α、JNK2β、ERK5、P38α、P38β、AMPK、STK11、SMARCA4、TP53、ESR1、GATA3、CDK2、SMAD1、NOTCH1、MYB、MYC、SMAD2、SMAD3、SMAD4、PRKACA、NLK或其任何组合。每种可能性是独立的实施方案。
在一些示例性实施方案中,PDM可以是KRas且FTR的靶部分可选自:ERK2、ERF、JNK和AKT1。每种可能性是独立的实施方案。
在一些示例性实施方案中,PDM可以是AKT2或AKT3且FTR的靶部分可选自:AKT1和RelA。每种可能性是独立的实施方案。
在一些示例性实施方案中,PDM可以是FGFR1且FTR的靶部分可选自:ERK2、JNK(诸如JNK1α1)、p38b、AKT1和STAT3。每种可能性是独立的实施方案。
在一些示例性实施方案中,PDM可以是BRaf且FTR的靶部分可选自:ERK2和ERF。每种可能性是独立的实施方案。
在一些示例性实施方案中,PDM可以是EGFR且FTR的靶部分可选自:ERK2、RelA、AKT1、p38b、JNK1a1。每种可能性是独立的实施方案。
在另一个实施方案中,本发明包括其中每种测定细胞都表达PDM和/或FTR的测定细胞。在另一个实施方案中,本发明包括其中每种测定细胞表达不同的PDM和/或FTR和/或PDR的测定细胞。在另一个实施方案中,本发明包括其中每种测定细胞转染了不同的DNA片段,其中各DNA片段编码不同的PDM和/或FTR的测定细胞。在一些实施方案中,测定细胞被置于/平铺/种植在具有指定位点(位置)的固体基底上。在一些实施方案中,针对每个位点的测定细胞是相同的。在一些实施方案中,针对每个位点的测定细胞是不相同的。在一些实施方案中,测定细胞被添加至每个位点的培养基中。在一些实施方案中,细胞被加至已具有在其上脱水的DNA构建体的固体基底。在一些实施方案中,先将细胞平铺在固体基底上并在预定的时间段之后转染。
在另一个实施方案中,本发明包括其中每种测定细胞转染了不同的DNA片段,其中各DNA片段编码不同的PDM和/或FTR和/或PDR的测定细胞。
在一些实施方案中,使用蛋白测定、结合测定、免疫测定、显微镜成像或本领域技术人员已知的任何其他合适的测定进行FTR定位的鉴定。
在一些实施方案中,本发明还包括检测测定细胞的形态变化的步骤。在一些实施方案中,本发明的方法不要求对任何患者的DNA测序。
根据一些实施方案,药物是抗癌药物。在一些示例性实施方案中,药物可以选自,但不限于:阿法替尼、Brentuximab vedotin、Buparlisib、卡博替尼(cabozantinib)、卡非佐米(carfilzomib)、西妥昔单抗(cetuximab)、克唑替尼(crizotinib)、达拉非尼(dabrafenib)、达沙替尼(dasatinib)、狄迪诺塞麦(denosumab)、厄洛替尼、依维莫司、吉非替尼(gefitinib)、替伊莫单抗(ibritumomab tiuxetan)、依鲁替尼(Ibrutinib)、艾代拉利司、甲磺酸伊马替尼(imatinib mesylate)、易普利姆玛(ipilimumab)、拉帕替尼、来那替尼、盐酸尼洛替尼一水合物(nilotinib hydrochloride monohydrate)、obinutuzumab、奥法木单抗(ofatumumab)、帕尼单抗(panitumumab)、帕唑帕尼(pazopanib)、帕妥株单抗(pertuzumab)、帕纳替尼(ponatinib)、瑞戈非尼、Rituxan、索拉非尼(sorafenib)、司美替尼、舒尼替尼(sunitinib)、替西罗莫司、曲美替尼(trametinib)、曲妥珠单抗(trastuzumab)、凡德他尼(vandetanib)、维罗非尼、维莫德吉(vismodegib)、阿柏西普(ziv-aflibercept),以及其任何组合。每种可能性是独立的实施方案。
根据一些实施方案,可以以任何期望的量/浓度将药物添加至受试细胞。例如,可以以以下浓度添加药物:1nM至1mM以及其任何子范围,诸如,例如,但不限于:150nM-750nM、12.5μM-100μM、200nM-25μM等。
根据一些实施方案,可以添加药物并且将药物与受试细胞孵育持续任何期望的时间段,诸如例如,在以下范围内的时间段:10分钟至24小时。
根据一些实施方案,提供了用于诊断患者的癌症的试剂盒。在一些实施方案中,提供了用于鉴定肿瘤细胞中异常的细胞信号传送途径的试剂盒。在一些实施方案中,提供了用于鉴定患者特定致癌突变的试剂盒。在一些实施方案中,提供了用于确定/检测/鉴定患者特定致癌突变的药物反应的试剂盒。在一些实施方案中,提供了用于测量患者突变基因对药物疗法的反应/抗性。
在一些实施方案中,本发明提供了用于通过鉴定患者特定致癌突变诊断受试者的癌症或癌症分子谱(molecular cancer profile)的试剂盒。根据一些实施方案,试剂盒可用于预测治疗成功率或鉴定参与癌症的旁分泌或自分泌因子。在另一个实施方案中,试剂盒包括至少一个检测报告物基因的工具(means)。在另一个实施方案中,试剂盒包括用于检测标志物的工具。在一些实施方案中,试剂盒包含以下的一个或更多个:用于容纳核酸分子和/或受试细胞的基底或容器、用于进行检测/易位测定的说明书、受试细胞、转染试剂或其任何组合。
如果期望的话,本发明的诊断组合物可以成品(article of manufacture)存在,例如,可包含诊断试剂和使用说明书的试剂盒,诸如FDA批准的试剂盒。所述试剂盒还可适于在容器上以规范药物生产、应用或销售的政府机构规定的形式附上公告,所述公告反映了组合物的形式或人或兽医用途被该机构批准。
在另一个实施方案中,本发明的方法和试剂盒增加癌症患者的存活率。本发明的测定非常适于试剂盒的制备。这类试剂盒可包含承载装置,所述承载装置被分区以在紧密布置的限定空间容纳一个或更多个容器装置,诸如小瓶、管、板、载玻片等,每个容器装置包含细胞测定的单独元件。
在一个实施方案中,提供了用于诊断受试者中的癌症的包括一组测定细胞的试剂盒,每种测定细胞包括本发明的不同蛋白,该试剂盒包括具有编码PDM(源自患者的生物样品)和/或FTR和/或FTR的核酸分子的基底以及用于反应测量和/或检测易位事件的打印的说明书,其中该基底还能够容纳测定细胞和从疑似患有癌症的人受试者分离的生物样品。
在一些实施方案中,转染的测定细胞在允许重组蛋白或标记的蛋白的表达的有效条件下培养。在一个实施方案中,本发明的标记的蛋白或标志物蛋白(例如PDM、FTR)是重组蛋白或嵌合体。在一些实施方案中,有效的培养条件包括,但不限于,允许蛋白表达的有效的培养基、生物反应器、温度、CO2、pH和氧气条件。在一个实施方案中,有效的培养基指细胞在其中被培养以产生本发明的重组多肽的任何培养基。在一些实施方案中,培养基通常包括具有可吸收的碳、氮和磷酸盐来源,以及适当的盐、矿物质、金属和其他营养物,诸如维生素的水溶液。在一些实施方案中,本发明的细胞可在常规发酵生物反应器、摇瓶、试管、微量滴定皿和培养皿中培养。在一些实施方案中,在重组细胞适合的温度、pH和氧含量下进行培养。在一些实施方案中,培养条件在本领域的普通技术人员的专业知识内。
在一些实施方案中,本发明利用再分配技术来监测并记录蛋白易位事件。在另一个实施方案中,用绿色荧光蛋白或其他荧光蛋白标记蛋白靶标,并产生被稳定转染或瞬时转染的细胞系。在另一个实施方案中,本发明的测定使用高通量、基于光学显微镜的仪器读取。
在另一个实施方案中,本发明的蛋白易位测定是高含量、高通量测定,主要用于先导系列的剖析(profiling of lead series)、作为细胞培养基成分的源自生物样品的PDM的初步筛选。在另一个实施方案中,本发明的蛋白易位测定包括使用Spinning Disc technology或任何其他基于显微术的技术的活细胞成像。
在一些实施方案中,拓扑定位技术(toponomic localization technique)用于跟踪并记录蛋白易位事件。在一些实施方案中,对本发明的蛋白利用了免疫荧光手段。在一些实施方案中,本发明的蛋白用荧光标志物标记。在一些实施方案中,对共聚焦显微镜图像进行评估和处理。在另一个实施方案中,标准数据集包括每种生物条件每种细胞的2-40个图像。在另一个实施方案中,进行自动化图像分析。在另一个实施方案中,自动化图像分析包括细胞区室或结构识别。
在另一个实施方案中,以不同的维度捕捉空间关系。在另一个实施方案中,进行蛋白标志物在有界区域中的浓度的定量评估。在另一个实施方案中,基于测量和评价像素点之间的距离的各向同性分布,本发明还提供了蛋白共定位研究。在另一个实施方案中,本发明提供了二维分析(区域)。在另一个实施方案中,本发明还提供了0维分析(点)。在另一个实施方案中,本发明提供了1维建模。
术语“包含(comprises)”、“包含(comprising)”、“包括(includes)”、“包括(including)”、“具有(having)”和其同源词意指“包括但不限于”。术语“包含(comprises)”和“包含(comprising)”在一些实施方案中分别限于“由...组成(consists)”和“由…组成(consisting)”。术语“由...组成(consisting of)意指“包括并局限于”。术语“基本上由...组成”意指组合物、方法或结构可包括另外的成分、步骤和/或部分,但仅当该另外的成分、步骤和/或部分不实质上改变请求保护的组合物、方法或结构的基本特征和新颖特征的情况下。
如本文所用的,除非上下文另外清楚指明,否则单数形式“一(a)”、“一(an)”和“该(the)”包括复数指代物。例如,术语“一个化合物(a compound)”或“至少一个化合物”可包括多个化合物,包括其混合物。
如本文使用的,述及本文提到的数值的术语“约”应理解为所提到的数值+/-10%。
如本文使用的术语“方法”指,用于完成给定任务的方式、手段、技术和程序,包括,但不限于,已知的方式、手段、技术和程序,或化学、药理学、生物学、生物化学和医学领域的从业者易于从已知的方式、手段、技术和程序开发的那些方式、手段、技术和程序。
在研究并非意在限制的以下实施例之后,本发明的另外的目的、优势、及新特征对本领域普通技术人员将变得明显。另外,如上文描写的以及以下权利要求部分要求保护的本发明的多个实施方案和多个方面中的每一个在以下实施例中均找到实验性支持。
以下实施例是为了更充分地说明本发明的一些实施方案而呈现。然而,其不应以任何方式被解释为限制本发明的宽泛的范围。
实施例
总体上,本文使用的命名法和本发明中利用的实验室程序包括分子学、生物化学、微生物学和重组DNA技术。这些技术已在文献中被充分描述。参见,例如,"Molecular Cloning:A laboratory Manual"Sambrook等,(1989);"Current Protocols in Molecular Biology"第I-III卷Ausubel,R.M.,编著(1994);Ausubel等,"Current Protocols in Molecular Biology",John Wiley和Sons,Baltimore,Maryland(1989);Perbal,"A Practical Guide to Molecular Cloning",John Wiley&Sons,New York(1988);Watson等,"Recombinant DNA",Scientific American Books,New York;Birren等(编著),"Genome Analysis:A Laboratory Manual Series",第1-4卷,Cold Spring Harbor Laboratory Press,New York(1998);如美国专利第4,666,828;4,683,202;4,801,531;5,192,659和5,272,057号中所列的方法学;"Cell Biology:A Laboratory Handbook",第I-III卷Cellis,J.E.,编著(1994);"Culture of Animal Cells-A Manual of Basic Technique"by Freshney,Wiley-Liss,N.Y.(1994),第三版;"Current Protocols in Immunology"第I-III卷Coligan J.E.,编著(1994);Stites等(编著),"Basic and Clinical Immunology"(第8版),Appleton&Lange,Norwalk,CT(1994);Mishell和Shiigi(编著),"Selected Methods in Cellular Immunology",W.H.Freeman和Co.,New York(1980);可用的免疫测定被广泛描述于专利和科学文献中,参见,例如,美国专利第3,791,932;3,839,153;3,850,752;3,850,578;3,853,987;3,867,517;3,879,262;3,901,654;3,935,074;3,984,533;3,996,345;4,034,074;4,098,876;4,879,219;5,011,771和5,281,521号;"Oligonucleotide Synthesis"Gait,M.J.,编著(1984);“Nucleic Acid Hybridization"Hames,B.D.,和Higgins S.J.,编著(1985);"Transcription and Translation"Hames,B.D.,和Higgins S.J.,编著(1984);"Animal Cell Culture"Freshney,R.I.,编著(1986);"Immobilized Cells and Enzymes"IRL Press,(1986);"A Practical Guide to Molecular Cloning"Perbal,B.,(1984)以及"Methods in Enzymology"第1-317卷,Academic Press;"PCR Protocols:A Guide To Methods And Applications",Academic Press,San Diego,CA(1990);Marshak等人,"Strategies for Protein Purification and Characterization-A Laboratory Course Manual"CSHL Press(1996);其全部通过引用并入本文。本文件贯穿全文提供了其他常规的参考文献。
生物样品收集
收集福尔马林固定的石蜡包埋(FFPE)的肿瘤活检物以及冷冻的新鲜肿瘤部分或活检物。FFPE样品用于提取已知参与癌症进展的特定的基因组外显子(诸如cKit外显子11)。新鲜的(新鲜的或新鲜冷冻的)活检物用于mRNA提取和组织间液提取。
因此,回顾性和前瞻性样品均被采集。回顾性研究基于来自已知治疗功效的病例的冷冻肿瘤切片。
前瞻性研究基于在手术/活检/支气管镜检后立即收集的新鲜或速冻的样品/活检组织/肿瘤切片。这使得扩增所有相关的测试蛋白(诸如癌基因或指示物)成为可能。其他体液诸如血浆样品(使用硫酸肝素凝胶管)、血液样品、腹膜积液、胸膜积液和通过支气管镜检获得的肺液是非常重要的,因为他们积累了许多肿瘤分泌物并且也被收集。
在肿瘤切除术(手术、活检、支气管镜检)后,将肿瘤组织放置于冰上的无菌袋或管中(未用福尔马林处理)。病理学从业者将肿瘤分割(考虑活的肿瘤部分的尺寸和位置)成需要固定的部分和最能代表肿瘤的部分,这些部分在冰上被新鲜递送以便进一步分析。病理学从业者找出富含恶性细胞且具有减少的量的基质或其他非恶性组织的组织部分或区域,并将其切下。如果组织的净重超过1克,则将组织进一步切成若干片,并放置在纤维素柱上并以100g离心10min(预期从每克组织得到100±50微升IF)。然后将组织转移至另一个15/50ml管并冷冻于-80℃冰柜。称为组织间液(IF)的离出液被冷冻在原管中。
穿刺活检-将组织放置于15圆锥管中并冷冻于-80冰柜。
经由支气管镜检的活检-将组织放置于15圆锥管中并冷冻于-80冰柜。
支气管肺泡灌洗-将提取液分在2个50ml falcon管中并离心(3000RPM,15min)。将液体转移进新管中,并将液体和细胞(原管中)均冷冻在80℃下。
胸膜积液-离心胸膜积液(3000RPM,15min.),将液体转移进新管,并将液体和细胞(原管中)均冷冻于-80℃下。
冷冻切片-如果可能的话,将肿瘤冻结在切片机(microtome)中并切片。
从福尔马林固定包埋的肿瘤活检物提取基因组DNA
为鉴定在特定外显子中存在已知突变的基因,诸如cKit在外显子11中的突变,EGFR外显子19、20,HER2外显子20,使用从FFPE组织提取的DNA。为此目的,使用标准的DNA提取试剂盒和操作说明(例如,Qiagen QIAamp DNA FFPE Tissue,目录号56404)。
扩增外显子并插入全长基因
为表达期望的外显子,从基因组DNA进行扩增并将外显子插入缺乏该外显子的全长基因中。为此目的,制备在表达备用载体(expression ready vector)中缺乏该外显子的全长基因,然后使用常规分子生物学技术将该外显子整合到构建体中。
新鲜的活检物:从冷冻的活检物提取所需量的组织
一部分活检物用于RNA纯化和组织间液提取。剩余的生物材料储存起来以备将来参考或其他分析(免疫组织化学(IHC)、FISH等)。
组织间液(IF)的提取
将按照下文详述地提取的组织间液(IF)储存起来供稍后用作被测试的细胞的激动剂以检测肿瘤细胞分泌并可赋予抗癌药物抗性的物质的存在。
IF提取通过在4℃下,在具有玻璃纤维过滤器的柱中以1500g离心组织样品7min进行。然后将来自柱底部的液体收集到新管中。
mRNA的提取
从样品提取的mRNA是扩增患者来源的标志物(PDM),即,已知为致癌基因并可能携带为细胞提供致癌性质的突变的基因(具有携带致癌突变的可能性的基因)所需要的。被测试的示例性基因包括:AKT1、AKT2、AKT3、ALK、BRAF、BRCA1、BRCA2、CBL、CTNNB1、EGFR、ERBB2、ERBB3、FGFR1、FGFR2、GNA11、GNAQ、HRAS、JAK2、KIT、KRAS、MET、NRAS、PDGFRA、PIK3CA、PTEN、RAF1、RET、ROS1、SMO、TP53、SMAD2、SMAD3、SMAD4、STAT1、STAT3、STAT5B、TGFBR2、FBXW7、MYC、LKB1、SMARCA4、TCF7L2、MAP3K1、AR、PR、ESR1、DDR2、MEK1和MEK2。
RNA提取通过包括胍盐-氯化铯法(Guanidium-Cesium Chlroride Method)、胍酸-酚法(Guanidium Acid-Phenol Method)的本领域已知的方法,和在离液盐(chaotropic salt)的存在下结合核酸的玻璃纤维过滤器进行,和/或通过使用商购可得的试剂盒进行(诸如Qiagen RNeasy试剂盒,目录号74106,根据制造商的说明书进行使用)。
cDNA的产生
为允许PDM的扩增,基于从组织提取的mRNA合成cDNA。基于模板mRNA,使用RNA-依赖性的DNA聚合酶逆转录酶,并使用寡-dT引物、随机六聚体引物(random hexameric primer)或特异性引物来合成cDNA。示例性方案包括使用SuperScriptTMIII First-Strand Synthesis SuperMix protocol(Life technologies,目录号18080-051)。
受试PDM的产生
受试PDM的产生以两个步骤进行:扩增所选择的PDM并附接其他元件以允许其在测定细胞中适当表达。
在一个直接的方法中,进行包含与被扩增的受试PDM相关的寡核苷酸的初步PCR反应以允许这些所选择的基因过多出现在cDNA样品内。在一个间接的方法中,基于特定的受试PDM的序列人工合成受试PDM,并且该人工PDM可以用作用于使用恰当的引物的PCR反应的模板。
在一些实例中,针对每个待扩增的基因,cDNA样本被分到单独的孔/管中。
使用为各个PDM设计的引物进行PCR反应以从cDNA文库扩增所选择的PDM基因,或基于人工产生的模板PDM使用为各个PDM设计的引物进行PCR反应。
以下组的引物用于以下被测试的PDM的PCR扩增(表1):
表1
在扩增PDM基因区域后,进行第二PCR反应以向各个PDM基因序列的5`末端添加启动子(或组成型启动子诸如CMV或诱导型启动子诸如四环素启动子)及向3`末端添加IRES接着是荧光报告物基因(诸如GFP、RFP、BFP或任何其他报告物基因,如指定的)。
在一些实例中,启动子和IRES+荧光报告物元件的添加通过分子生物学克隆工具,通过借助PCR方法、连接酶或重组方法(诸如分别为T4 DNA连接酶或InFusion酶(Clontech))将PCR产物融合至期望的元件进行。
当全长核酸分子(即5'启动子-PDM-3'IRES+报告物(或这些元件的任何其他顺序))形成时,进行使用PCR反应的扩增,以获得足量的用于转染细胞的核酸分子。
在一些个例中,核酸分子的扩增通过将全长核酸分子连接到适当的表达载体上并转化到细菌中来实现。使用标准质粒提取试剂盒,诸如Qiagen QIAprep Miniprep试剂盒提取由此形成的质粒。在一些个例中,各种PDM的线性PCR片段用于转染进入受试细胞中。
FTR的生成:
以下组的引物用于PCR扩增以下FTR的靶部分(表2):
表2
表达构建体(FTR和PDM的混合物)的转染
根据预先设计的矩阵,将用于分析的按上文描述的制备的每个报告物基因(FTR)与对照野生型PDM基因或受试PDM基因混合,并与适当的转染试剂混合。
在一种可选方案中,将转染混合物放置在适当的固体支持基底上并任选地在该适当的固体支持基底上脱水。在各种设置中,基底包括多种固体基底,诸如:显微镜载玻片、芯片、细胞培养板、多孔板、96孔板、384孔板等。将各种混合物放置在指定的、可追踪的位点/点(即,指定的孔或载玻片或芯片上的指定位置)中。对基底的转染混合物,将处于正常的全生长培养基中的固定数目的细胞(在约100至100,000的范围内,取决于以上所述的基底类型)分配至各个点上。基于所测试的PDM和测定方法,细胞选自HeLa细胞、HEK 293细胞、NCI60细胞系诸如A549、EKVX、T47D、HT29或任何其他合适的细胞系。将受试细胞放置在固体基底上,并根据细胞的类型、生长培养基和转染条件孵育12-48小时。孵育时间允许细胞粘附至基底,并允许细胞导入和表达FTR和PDM。
在另一种可选方案中,根据预先设计的矩阵将细胞平铺在固体基底上(在指定的、可追踪位点/点(即,指定的孔或载玻片或芯片上的指定位置))。在预定的时间段之后,在适当的转染条件用FTR和适当的PDM(WT PDM或受试PDM)转染细胞。FTR和合适的PDM可位于两个不同的分子上,或位于编码这两个基因的一个分子上。
测定实施:诱导型启动子
在报告物FTR充分表达之后,将生长培养基替换为低血清培养基(以除去培养基中存在的任何生长因子/配体)以减至最少的背景刺激的信号传送。
当信号传送水平显著减少(4至16小时内)时,启动PDM的诱导表达。这是通过添加四环素(当使用四环素诱导型启动子时)和蜕皮激素(当使用蜕皮激素诱导型启动子时)实现的。
在一些实例中,添加组织间液(IF)和/或抗癌药物以诱导PDM的表达,从而测试IF或药物对PDM的影响。
测定实施-组成型启动子
在细胞充分表达FTR和PDM(均在组成型启动子的控制下)后,将生长培养基替换为低血清培养基(以除去培养基中存在的任何生长因子/配体),以减至最少的背景刺激的信号传送。
在一些实例中,添加组织间液和/或抗癌药物以诱导PDM的表达,并从而测试IF或药物对PDM的影响。
图像采集和分析
在PDM表达(转染30小时后)后,通过磷酸盐缓冲的盐水(PBS)洗涤3次,在4%多聚甲醛(PFA)中孵育5分钟,并随后用PBS洗涤3次固定细胞。然后用盖玻片覆盖载玻片,并对每个相应的FTR的定位成像。
对对照野生型细胞以及PDM转染的细胞中的每种FTR进行图像分析并做比较。将FTR在对照细胞与PDM转染的细胞中的定位之间的差异定量,并使用所述差异确定所测试的PDM的致癌形式或野生型形式是否存在于被测试的样品中。定量使用标准图像分析软件,诸如ImageJ进行。
使用HeLa细胞作为测定细胞的示例性测定:
第0天:在室温(RT)下用0.01%的聚-l-赖氨酸预包被载玻片5分钟,然后用无菌水(DDW)洗涤。将水吸掉并将载玻片干燥2小时。将HeLa细胞(15000个细胞)平铺在每个孔的200μl完全培养基(完全培养基:DMEM、10%FBS、1%青霉素/链霉素(P/S))中。
第1天:将转染试剂(FugeneHD试剂(Promege,目录号E2311)加热至RT并涡旋。对于每个孔,在管中制备转染混合物,所述转染混合物包括:管中的50/100/200ng PDM表达构建体;50/100ng合适的FTR的表达构建体;Optimem缓冲液(至总计10μl)和FugeneHD(对于每3μg DNA为1μl)。在RT下孵育转染混合物15分钟。从孔中吸出细胞培养基,并对每个孔补充100μl转染培养基(DMEM、10%FCS、无抗生素)。将10μl转染混合物添加至每个孔中。然后将细胞孵育在37℃的湿润培养箱(5%CO2)中。6-8小时后,培养基被更换为饥饿培养基1(具有0.1%FCS、1%青霉素/链霉素的DMEM),并将细胞孵育在37℃、5%CO2的湿润培养箱中。对于需要药物/化学抑制剂的24小时孵育的测定,所述药物/化学抑制剂以所需浓度被添加。
对于需要与药物孵育的测定:根据需要用补充有药物的饥饿培养基2替换培养基。然后将细胞孵育在37℃的湿润培养箱(5%CO2)中。另外,如果需要更短的药物孵育时间,其可以被进行。
第2天:26小时后(即,固定细胞之前4小时),将培养基换成饥饿培养基2(具有1%P/S的DMEM)。然后将细胞孵育在37℃的湿润培养箱(5%CO2)中。
对于需要诱导信号传送的测定:根据需要用补充有诱导物的饥饿培养基2替换培养基。然后将细胞孵育在37℃的湿润培养箱(5%CO2)中。另外,如果需要更短的药物/化学抑制剂孵育时间,其可以被进行。
转染30小时后,通过以下方法固定细胞(所有步骤在室温下):将细胞用PBS洗涤3次。用固定溶液(PBS中的5%葡萄糖/4%多聚甲醛(PFA))固定持续10分钟,用PBS洗涤3次。将细胞任选地用DAPI溶液染色,之后用PBS洗涤细胞三次。
实施例1:患者BRAF突变赋予对ERBB2抑制剂来那替尼的抗性。
一名66岁男性被诊断为患有转移性肺腺癌。患者用赫塞汀治疗,并且随后用来那替尼治疗,这未提供无进展生存期(progression free survival)(PFS),并且进展性疾病维持。患者在治疗2个月后去世。除了ERBB2/HER2致癌突变(A771_Y772insYVMA,G776C)和AKT1扩增以外,还鉴定了BRAF I554T突变,并且使用本文公开的方法证明BRAF I554T突变是高度致癌的。将来自肝转移性部位的针刺活检物用作生物样品。
用WT BRAF或患者的突变的BRAF与相应的FTR,ERK2 GFP一起转染Hela测定细胞。将细胞放置不处理或者用600nM浓度的ERBB2抑制剂来那替尼处理持续6小时。转染30小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量细胞核(N)与细胞质(C)中FTR强度之间的比率(N:C比率)。结果在图3A-B中示出,该结果显示与WT基因相比,当突变的患者基因被转染入细胞时观察到ERK的较高的N:C。如图3A中进一步所示,来那替尼处理表达WT BRAF或者患者突变体BRAF的细胞,导致ERK易位到细胞核的微小减少,表明受试药物缺乏抑制活性,因为该活性依然显著高于当WT形式在细胞中表达时所观察到的活性。
实施例2:患者BRAF突变对瑞戈非尼敏感。
如在实施例1中的,用WT BRAF或患者的突变的BRAF(I554T)与相应的FTR,ERK2 GFP一起转染Hela测定细胞。将细胞放置不处理或者用1μM浓度的BRAF抑制剂瑞戈非尼处理持续24小时。转染30小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量细胞核(N)与细胞质(C)中FTR强度之间的比率(N:C比率)。结果在图4A-B中示出,该结果显示与WT基因相比,当突变的患者基因被转染入细胞时观察到ERK的较高的N:C。如图4A中进一步所示,当测量患者突变体对直接抑制剂瑞戈非尼的反应时,突变体的致癌活性被消除并且回到比当表达WT形式时观察到的那些活性更低的水平。瑞戈非尼还显著抑制WT信号传送作用。
实施例3:患者EGFR突变对厄洛替尼有抗性但是对阿法替尼敏感
一名34岁女性被诊断为患有IV期肺腺癌。用厄洛替尼与化学疗法一起治疗患者,并且反应良好。12个月后发展为对治疗的抗性。这随后进行阿法替尼治疗,具有良好反应。该反应短暂(4个月),并且之后进展对厄洛替尼或阿法替尼无反应。在出现抗性后,通过在若干基因中测序(NGS)鉴定以下突变:MUTYH(p.V376L;.V390L;p.V362L;.V363L;p.V387L);EGFR(p.G719A);EGFR(p.T790M);EGFR(p.L861Q);TRIM24(p.C595S;p.C629S);BRD3(p.G677W);NOTCH1(p.C222fs);BIVM-ERCC5;ERCC5(p.M254V;p.M708V)。
使用本文公开的方法的先前实验鉴定了患者的EGFR突变为高度致癌的,影响多种途径。EGFR中的三重突变由2个已知的致癌突变G719A和L861Q,以及赋予对若干EGFR抑制剂的抗性的第3个致癌突变(T790M)组成。该突变已经被证明为EGFR中的继发突变,由于该突变干扰厄洛替尼和吉非替尼的结合位点而消除了厄洛替尼和吉非替尼的作用。
用WT EGFR或患者的突变的EGFR(EGFR G719A/T790M/L861Q)与相应的FTR,ERK2GFP一起转染Hela测定细胞。将细胞放置不处理或者用300nM浓度的厄洛替尼(患者已发展对其的抗性的EGFR抑制剂)处理持续24小时,或者用阿法替尼(第二代EGFR抑制剂,其在患者中表现出一些反应)处理。转染30小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量细胞核(N)与细胞质(C)中FTR强度之间的比率(N:C比率)。结果示于图5A-B中。患者EGFR中存在的突变具有已知功能,因此测试其对特定靶向疗法的反应是否会类似于在临床中观察到的结果(即,对厄洛替尼有抗性并且对阿法替尼敏感)。如图5A中所示,厄洛替尼处理有效力地抑制WT EGFR的活性水平,但是对患者三重突变体EGFR没有作用。这是由于已知赋予对该抑制剂的抗性的T790M突变引起的。这与在用该药物治疗后患者中所报告的抗性一致。相比之下,如图5B中所示,患者对其表现出良好反应的EGFR抑制剂阿法替尼,在测定系统中能够有效力地抑制三重突变体的EGFR的致癌活性。尽管该活性水平未反转到WT水平,但是活性降低显著。
总之,本文所示的结果表明,本文公开的方法事实上能够以高度显著性预测被测试的两种不同靶向疗法药物的抗性和敏感性。此外,图5A-B所示的结果还与临床中观察到的结果一致,为这些药物的效率提供了机制性解释。因此,这些结果示例了公开的方法和系统鉴定多种突变的药物反应以及在多种药物对相同靶标的情况下允许药物选择的能力。
实施例4:ERK1/2途径的亚细胞易位测定能够辨别BRAF对不同的单一靶向疗法与多重靶向疗法的反应,并且鉴定BRAF抗性/敏感性突变。
用WT BRAF或突变的BRAF与相应的FTR,ERK2-GFP一起转染Hela测定细胞。细胞未处理或者用增加浓度的BRAF抑制剂维罗非尼(7.5uM-25uM,图6B)、瑞戈非尼(500nM-3uM,图6C)或MEK1/2抑制剂司美替尼(300nM-3uM,图6D)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的BRAF与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图6B-D中所示的结果显示,增加剂量的药物减少突变形式相比于WT基因的ERK易位的差异。该作用被证明是突变特异性的,因为与V600突变相比非V600突变对不同的药物不敏感。如图6E和6F中进一步显示的,与单独每种药物相比,组合两种不同的药物(维罗非尼和司美替尼或者维罗非尼和瑞戈非尼)在途径抑制中未赋予额外的益处。
实施例5:ERK1/2和STAT途径的亚细胞易位测定能够辨别EGFR对不同的单一靶向疗法与多重靶向疗法的反应,并且鉴定EGFR抗性/敏感性突变。
用WT-BRAF或突变的BRAF与相应的FTR,ERK2-GFP或STAT3-GFP一起转染Hela测定细胞。细胞被放置未处理或者用增加浓度的EGFR抑制剂阿法替尼(625nM-25uM,图7B)或增加浓度的MEK1/2抑制剂司美替尼(300nM-30uM,图7C)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的EGFR与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图7B中所示的结果显示,增加剂量的药物减少突变形式中的一些相比于WT基因的ERK易位的差异。该作用被证明是突变特异性的,因为与L858R/T790M突变相比L858R突变体对药物敏感,可能是由于由T790M突变赋予的抗性。如图7C和图7D进一步示出的,测试同一药物司美替尼在两种不同的信号传送途径(ERK1/2和STAT)中的作用再次表明,不含T790M的突变体(L858R和L861Q)对MEK抑制剂敏感,而T790M突变无反应。重要的是,图7E和图7F显示,与单独每种药物相比,组合两种不同的药物(阿法替尼和司美替尼或者阿法替尼和瑞戈非尼)在途径抑制中引起额外的益处。结果表明,靶向沿EGFR-ERK1/2途径的多个靶点的药物组合比单独每种药物更有效,靶向多个靶点的药物组合甚至在含T790M的突变中有效。
实施例6:ERK1/2和STAT途径的亚细胞易位测定能够辨别ERBB2对不同的单一靶向疗法与多重靶向疗法的反应,并且鉴定ERBB2抗性/敏感性突变。
用WT ERBB2或突变的ERBB2与相应的FTR,ERK-GFP或STAT3-GFP一起转染Hela测定细胞。细胞被放置未处理或者用增加浓度的ERBB2抑制剂来那替尼(500nM-3uM,图8B)或拉帕替尼(500nM-3uM,图8C),或者MEK1/2抑制剂司美替尼(500nM-7uM,图8D)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的ERBB2与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图8B和图8C中所示的结果显示,增加剂量的ERBB2抑制剂减少V777L突变体中的STAT3易位的差异,但是未减少S310F突变中STAT3易位的差异。如图8D进一步示出的,测试MEK抑制剂司美替尼在ERK1/2途径中的作用显示,S310F突变对该药物敏感,而另一个突变V842I未受影响。最后,图8E和图8F显示,与除了S310F(图8F)以外的单独每种药物相比,组合两种不同药物(来那替尼和司美替尼或者拉帕替尼和司美替尼)在途径抑制中未引起额外的益处。这表明,靶向沿ERBB2-ERK1/2和ERBB2-STAT3途径的多个点的药物组合不比单独每种药物更有效。
实施例7:P38和NFkB途径的亚细胞易位测定能够辨别PIK3CA对不同的单一靶向疗法与多重靶向疗法的反应,并且鉴定PIK3CA抗性/敏感性突变。
用WT PIK3CA或突变的PIK3CA与相应的FTR,P38或REL-A GFP一起转染Hela测定细胞。细胞被放置未处理或者用增加浓度的PIK3CA抑制剂艾代拉利司(150nM-750nM,图9B)、依维莫司(0.5uM-4.5uM,图9C)、Buparlisib(150nM-1uM,图9D)或替西罗莫司(0.5uM-3uM,图9E)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的PIK3CA与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图9B中所示的结果显示,增加剂量的PIK3CA抑制剂增加V344A、Q546L和N345K突变体中REL-A易位的量级。类似地,图9C显示,增加剂量的PIK3CA抑制剂降低V344A和H1047R突变体中P38易位的量级,但是不影响N345K突变体。如图9D和图9E中进一步示出的,测试另外的PIK3CA抑制剂Buparlisib和替西罗莫司在P38途径中的作用,表明N345K和H1047R突变对第一种药物敏感(图9D)但对后者不敏感(图9E)。最后,图9F显示,与除了H1047R以外的单独每种药物相比,组合两种不同药物(艾代拉利司和依维莫司)在途径抑制中未引起额外的益处。这表明,靶向沿PIK3CA-P38途径的多个点的药物组合通常不比单独每种药物更有效,而取决于特定的突变。
实施例8:STAT3途径的亚细胞易位测定能够辨别cKIT对不同的单一靶向疗法与多重靶向疗法的反应,并且鉴定cKIT敏感性突变。
用WT cKIT或突变的cKIT与相应的FTR,STAT3GFP一起转染Hela测定细胞。将细胞放置不处理或者用增加浓度的cKIT抑制剂伊马替尼(200nM-25uM,图10B)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的cKIT与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图10B中所示的结果表明,增加剂量的cKIT抑制剂降低W557-K558del突变体中STAT3易位的量级,在200nM明显达到最大效果。此外,图10C显示,与单独每种药物相比,组合两种不同的药物(伊马替尼和司美替尼)在途径抑制中未引起额外的益处。
实施例9:ERK1/2途径的亚细胞易位测定能够辨别ERBB2和BRAF单独地或组合地对不同的单一靶向疗法与多重靶向疗法的反应。
用WT ERBB2或BRAF或者突变的ERBB2、突变的BRAF或者两者的组合与相应的FTR,ERK2-GFP一起转染Hela测定细胞。细胞被放置未处理(UT)或者用ERBB2抑制剂来那替尼、BRAF抑制剂维罗非尼或两种药物的组合(图11B)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的两种基因与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图11B中所示的结果显示,尽管单独每种药物具有在某种程度上抑制ERK1/2途径的能力,但是组合这两种不同的药物(来那替尼和维罗非尼)导致协同抑制,因为与每种药物当被单独提供时的作用相比,在途径抑制中观察到额外的益处。
实施例10:ERK1/2途径的亚细胞易位测定能够辨别EGFR和KRAS单独地或组合地对不同的单一靶向疗法与多重靶向疗法的反应。
用WT EGFR或KRAS或者突变的EGFR、突变的KRAS或者两者的组合与相应的FTR,ERK2-GFP一起转染Hela测定细胞。细胞被放置未处理(UT)或者用EGFR抑制剂阿法替尼、MEK1/2抑制剂司美替尼或两种药物的组合(图12B)处理持续18小时。转染24小时之后,将细胞固定并且利用荧光显微镜成像。对FTR在细胞质和细胞核中的量进行定量。测量WT形式的两种基因与不同突变体之间的细胞核(N)与细胞质(C)中FTR强度之间的比率差异(ΔN:C比率)。图12B中所示的结果显示,尽管单独每种药物具有在某种程度上抑制ERK1/2途径的能力,但是与每种药物当被单独提供时的作用相比,仅组合这两种不同的药物(阿法替尼和司美替尼)在途径抑制中引起额外益处。
具体实施方案的以上描述将如此充分地揭示本发明的总体性质,使得其他人通过应用现有知识,可容易地进行调整和/或修改这类具体实施方案以用于多种应用,而无需过多的实验且不背离一般概念,并且因此,这类调整和修改应该被理解为并且预期被理解为包含在所公开的实施方案的等价方式的含义和范围内。虽然已结合本发明的具体实施方案描述了本发明,但是显然地,对于本领域技术人员,许多替换、修改和变化将是明显的。因此,涵盖落入所附权利要求书的精神和宽范围内的所有这些替换、修改和变化是期望的。
序列表
<110> 诺威尔卢斯德克有限公司
<120> 用于确定患者特定致癌突变的药物反应的方法
<130> NNDX 004 PCT
<150> 62/026715
<151> 2014-07-21
<160> 47
<170> PatentIn version 3.5
<210> 1
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 1
atgagcgacg tggctattgt 20
<210> 2
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 2
tcaggccgtg ccgctggc 18
<210> 3
<211> 19
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 3
atggcggcgc tgagcggtg 19
<210> 4
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 4
tcagtggaca ggaaacgcac 20
<210> 5
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 5
atgcgaccct ccgggacg 18
<210> 6
<211> 24
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 6
tcatgctcca ataaattcac tgct 24
<210> 7
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 7
atgacggaat ataagctggt ggt 23
<210> 8
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 8
tcaggagagc acacacttgc 20
<210> 9
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 9
atgcccaaga agaagccgac 20
<210> 10
<211> 19
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 10
ttagacgcca gcagcatgg 19
<210> 11
<211> 23
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 11
atgactgagt acaaactggt ggt 23
<210> 12
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 12
ttacatcacc acacatggca 20
<210> 13
<211> 19
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 13
atggggactt cccatccgg 19
<210> 14
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 14
ttacaggaag ctgtcttcca cc 22
<210> 15
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 15
atgcctccac gaccatcatc 20
<210> 16
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 16
tcagttcaat gcatgctgtt 20
<210> 17
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 17
atgacagcca tcatcaaaga ga 22
<210> 18
<211> 25
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 18
tcagactttt gtaatttgtg tatgc 25
<210> 19
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 19
atggagcaca tacagggagc 20
<210> 20
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 20
ctagaagaca ggcagcctcg 20
<210> 21
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 21
atggaggagc cgcagtca 18
<210> 22
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 22
tcagtctgag tcaggccctt 20
<210> 23
<211> 25
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 23
atgaatgagg tgtctgtcat caaag 25
<210> 24
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 24
tcactcgcgg atgctgg 17
<210> 25
<211> 21
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 25
atgagcgatg ttaccattgt g 21
<210> 26
<211> 22
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 26
ttattctcgt ccacttgcag ag 22
<210> 27
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 27
atgtggagct ggaagtgc 18
<210> 28
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 28
tcagcggcgt ttgagtc 17
<210> 29
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 29
atggtcagct ggggtcg 17
<210> 30
<211> 25
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 30
tcatgtttta acactgccgt ttatg 25
<210> 31
<211> 27
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 31
gcctgctgaa aatgactgaa tataaac 27
<210> 32
<211> 31
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 32
ttacataatt acacactttg tctttgactt c 31
<210> 33
<211> 21
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 33
atgtcgtcca tcttgccatt c 21
<210> 34
<211> 21
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 34
ttatgacatg cttgagcaac g 21
<210> 35
<211> 16
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 35
atggcggcgg cggcgg 16
<210> 36
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 36
ttaagatctg tatcctgg 18
<210> 37
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 37
atgaagaccc cggcggacac 20
<210> 38
<211> 19
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 38
tcaggagtct cggtgctcc 19
<210> 39
<211> 17
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 39
atgagcagaa gcaagcg 17
<210> 40
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 40
tcactgctgc acctgtgc 18
<210> 41
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 41
atggacgaac tgttccccct 20
<210> 42
<211> 21
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 42
taggagctga tctgactcag c 21
<210> 43
<211> 14
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 43
atgtcgggcc ctcg 14
<210> 44
<211> 20
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 44
tcactgctca atctccaggc 20
<210> 45
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 45
atggcccaat ggaatcag 18
<210> 46
<211> 18
<212> DNA
<213> 人工序列
<220>
<223> 引物
<400> 46
tcacatgggg gaggtagc 18
<210> 47
<211> 6
<212> PRT
<213> 人工序列
<220>
<223> 肽
<400> 47
Cys Cys Pro Gly Cys Cys
1 5
- 还没有人留言评论。精彩留言会获得点赞!