用于薄膜制造和催化的新型N‑氨基胍基金属配合物的制作方法
本专利申请涉及具有至少一个n-氨基胍基配体的新型金属配合物和所述金属配合物的制备及其用途。
所述新型金属配合物优选地用作薄膜工艺中的前体。具体而言,所述新型金属配合物用作通过气相沉积工艺诸如cvd(化学气相沉积)、mo-cvd(金属有机化学气相沉积)和ald(原子层沉积)产生功能层的前体。此外,所述配合物优选地也适合作为烯烃氢胺化和烯烃聚合的催化剂。
化学气相沉积(cvd)是一种气相反应(通常在基底表面上或者基底表面附近)。在此类反应中,将反应气体传送进入反应室的同时基底被涂覆。通常被预热的气体被加热的基底热激活并且它们相互反应。在该反应的过程中,期望的材料被沉积和化学键合(化学吸附)。
除了操作压力和其他操作参数不同的数不清的cvd变型之外,还存在代表cvd工艺被较大或较小程度改进的某些涂覆工艺:
在已知为等离子体聚合的工艺中,通过等离子体激发的气态单体在基底上形成高度交联的层。
原子层沉积(ald)是一种高度改进的cvd工艺,其中在表面上的反应或吸附在表面被完全覆盖之后自动停止。这种自限制反应要运行多个周期(在其间是冲洗步骤),从而获得非常好的纵横比(长度/厚度比)和精确的层厚度。
n-氨基胍基配体的分类基于胍骨架,其中三个胍氮原子中的至少一个通过直接n-n键合被nr2基团取代。在配位状态,配体是二齿的,其中两个n原子配位至金属(m)并且形成序列m-n-n-c-n-的五元环。在配位状态,配体优选地具有单负电荷。可以形成均配和杂配金属配合物。
包含二齿n-氨基胍配体的金属配合物从文献中已知。
santra等人在eur.j.inorg.chem.,2002,1124-1131中描述了2-(芳基偶氮)嘧啶(aapm)的钯配合物的制备及其与芳基胺的偶联反应。描述的pd(aapm)cl2配合物具有式a,
其中r是h、ch3或cl。
所述配合物包含序列pd-n-n-c-n-的五元环。然而,与胍核心结构键合的氮原子被大的芳基基团取代。从而,配合物的挥发性强烈地减小。而且,两个胍氮原子通过c3-单元相互连接,使得在包括胍核心的配体内形成了六元环。这导致柔性下降以及受损的和重复性较差的分解。
pratihar等人在inorganicachimicaacta,2010,363,831-840中描述了二氯-(1-烷基-2-(萘基-β-偶氮)咪唑)钯(ii)配合物(pd(β-nair)cl2)与芳基胺的偶联反应。描述的配合物显示在式b中,
其中r是ch3、ch2ch3或ch2ph。
该描述的配合物包含序列pd-n-n-c-n-的五元环。然而,与胍核心结构键合的氮原子被大的芳基基团取代。从而,配合物的挥发性强烈地减小。而且,两个胍氮原子通过c2-单元相互连接,使得在包括胍核心的配体内形成了五元环。这导致柔性下降以及受损的和重复性较差的分解。
wo2012/113761公开了n-氨基脒基配合物及其用途。然而,从这些配体没有获得镧系配合物,并且获得的配合物难以纯化,这是因为它们的低熔点使得重结晶更加困难。
在半导体组件(处理器、存储器芯片、传感器芯片等)的制造中,常常应用cvd、mo-cvd和ald工艺沉积金属层、氧化层和氮化层。现在这些工艺在半导体技术和微电子方面获得了巨大的重要性。
在这些工艺中,经常在存在反应性气体(例如氢气、氨气或1,1-二甲基肼)下,通过蒸发和加热合适的前体化合物在分解点以上在基底和气相之间的界面处涂覆基底。这些类型的工艺例如用于生成gan、inn、tan、tin或si3n4的层。
也可能沉积金属层(例如pd、ru或co)。为了适合用于cvd、movpe和ald,适合的配体和金属配合物应当具有分子结构(并且理想地应当作为单体存在)、具有低的摩尔质量并且在储存温度以上的温度下具有高的挥发性和低的分解点。
而且,它们在室温下应当是热稳定的,这样在沉积工艺之前不存在分解。此外,化合物应当具有均匀的、可重复的分解机制并且对于分子中的分裂应当具有合适的预定破坏点。最后,对于在相同cvd条件下的限定的前体化合物,应该总是可以以一致的质量沉积同样的层。
此类前体化合物的合适的配体应该提供金属中心的良好空间屏蔽、是富电子的并且应该电子上满足金属中心,从而降低路易斯酸度和抑制化合物聚集为低挥发性的配位聚合物。而且,在分解期间,金属中心的还原通常是必要的。包含高比例肼结构单元的配体,例如本专利申请的配体,它们本身携带有还原等效物。
优选地,用于制造半导体组件的前体化合物的配体具有高的氮含量。一方面,配体中高的氮含量提高了氮并入半导体元件的比率。另一方面,配体中高的氮含量使得反应性气体的使用受阻,因为它们是分子内地包含在配体中并因此它们在配体分解后产生。
已有的金属配合物例如具有n-氨基胍配体的那些具有缺点。例如,它们具有大的芳基基团键合至氨基基团,胍核心被其取代。这导致高的摩尔质量并因此导致低的挥发性。而且,两个胍氮原子是配体间环结构的部分,其导致配体降低的构象柔性。因此这些氨基胍基配合物用作薄膜沉积工艺的前体可能导致重复性、层质量、沉积率和产率方面的缺陷。
因此,目的是提供改进的金属配合物。这些金属配合物应当适合用于薄膜沉积工艺。它们应当具有低的摩尔质量和高的挥发性。它们也应当是储存稳定的。它们应当可以以限定的和可重复的方式分解。所述金属配合物也应当适合用作烯烃氢胺化和烯烃聚合的催化剂。
通过在本权利要求书中要求保护的新型n-氨基胍基金属配合物实现了该目的。这些新型金属配合物的特征在于它们包含至少一个n-氨基胍基配体。这些金属配合物具有低的摩尔质量和有利地高的挥发性。它们的特征还在于高的氮含量,其高于例如n-氨基脒基配合物中的氮含量。
标题化合物,特别是结合并入了金属烷基或金属氢化物官能团的第13族金属(b、al、ga、in)和n-氨基胍基配体的那些的应用领域是稀释氮化物iii-v半导体材料例如gaas1-xnx或inp1-xnx的金属有机气相外延(movpe)生长。目标是降低在将大量的n原子通过movpe流延到gaas和inp生长工艺中和一般地到亚稳定的iii-v半导体材料中期间通常需要的高的活化壁垒、高的沉积温度和大过量的氨气或其他氮源。与相应的n-氨基脒基配合物相比更高的氮含量邻近直接金属-氮键,可导致更高比率的氮移植入沉积的层以及与n-氨基脒基化合物相比不同的配体分解途径。由于具有特别的弱n-n键的一个或两个肼单元与金属键合,这些新型的前体特别适合用于薄膜沉积工艺。本专利申请因此也涉及该金属配合物作为薄膜工艺中的前体的用途,特别是其中薄膜工艺选自cvd工艺,例如mo-cvd工艺和ald工艺。
而且,新型金属配合物也适合作为烯烃氢胺化和烯烃聚合的催化剂。由于其配体是有利地富电子的,该金属配合物能够提高烯烃氢胺化和聚合反应的催化活性。本专利申请因此也涉及金属配合物作为烯烃氢胺化或烯烃聚合的催化剂的用途。
本专利申请涉及具有至少一个n-氨基胍基配体的金属配合物,其中这些金属配合物具有式1a或1b
其中
m是选自元素周期表(pte)第1-15族、镧系或锕系的金属;
r1是氢,具有至多5个碳原子的环状、直链或支链烷基,nh2,nh(ch3),n(ch3)2,n(c2h5)2或n-吡咯烷基,或r1和r2与它们键合的氮原子一起形成吡咯烷环;
r2是氢,具有至多5个碳原子的环状、直链或支链烷基,或r2和r1与它们键合的氮原子一起形成吡咯烷环;
r3是氢,或具有至多8个碳原子的环状、直链或支链烷基,nh2,nh(ch3),n(ch3)2,n(c2h5)2或n-吡咯烷基或sime3基团;
r4和r5相互独立地是氢或具有至多4个碳原子的直链或支链烷基,或r4和r5与它们键合的氮原子一起形成吡咯烷环;
x是单阴离子共配体,选自:氢阴离子(h-),卤素,具有至多8个碳原子的环状、直链或支链烷基(alkylide),具有至多10个c原子的取代或未取代的芳基(arylide)和杂芳基,烷氧基配体,烷硫基或烷砷基配体,或者叔氨基配体;
y是双阴离子共配体,选自:氧基[o]2-,硫基[s]2-或亚氨基[nr6]2-,其中r6是具有至多8个碳原子的环状、支链或直链烷基或者是具有至多20个碳原子的取代或未取代的芳基;
l是中性的2电子供体配体;
a是1和4之间的整数,并且
n、m和p各自相互独立地是0、1、2、3或4。
在本专利申请的配合物中,金属原子可以以+1至+6的标准氧化态存在。合适的氧化态特别地选自+1、+2和+3。至少一个n-氨基胍基配体携带负电荷并且因此为单阴离子形式。因此,配体是富电子的并且电子上满足金属中心。从而,配体有利地降低路易斯酸度并抑制聚集成低挥发性的配位聚合物。这被证明对于薄膜沉积工艺是有利的。
配合物中使用的中心原子m是元素周期表(pte)第1-15族的金属。这包括pte的s区的金属(第1和2族,即碱金属和碱土金属)、p区金属(第13、14和15族)和d区金属(第3-12族过渡金属)。该限定也包括pte周期内的所有金属,因此包括贵金属。中心原子m也可选自镧系或锕系。已知镧系是金属镧(la)、铈(ce)、镨(pr)、钕(nd)、钷(pm)、钐(sm)、铕(eu)、钆(gd)、铽(tb)、镝(dy)、钬(ho)、铒(er)、铥(tm)、镱(yb)和镥(lu)。已知锕系是金属锕(ac)、钍(th)、镤(pa)、铀(u)、镎(np)、钚(pu)、镅(am)、锔(cm)、锫(bk)、锎(cf)、锿(es)、镄(fm)、钔(md)、锘(no)、铹(lr)。
优选地,m选自硼(b)、铝(al)、镓(ga)、铟(in)、硅(si)、锗(ge)、锡(sn)、砷(as)、锑(sb)、钛(ti)、锆(zr)、铪(hf)、铬(cr)、锰(mn)、铁(fe)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钌(ru)、铑(rh)、钯(pd)和铂(pt)。更优选地,m选自硼(b)、铝(al)、镓(ga)、铟(in)、锗(ge)、锡(sn)、锑(sb)、钛(ti)、锆(zr)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钌(ru)、铑(rh)、钯(pd)和铂(pt)。甚至更优选地,m选自硼(b)、铝(al)、镓(ga)、铟(in)和钯(pd)。
在具体的实施方案中,配合物具有式1a并且配合物中使用的中心原子m是元素周期表(pte)第1-15族的金属。具体地,中心原子m选自pte的s区的金属(第1和2族,即碱金属和碱土金属)、p区金属(第13、14和15族)和d区金属(第3-12族过渡金属)。更具体地,m选自硼(b)、铝(al)、镓(ga)、铟(in)、硅(si)、锗(ge)、锡(sn)、砷(as)、锑(sb)、钛(ti)、锆(zr)、铪(hf)、铬(cr)、锰(mn)、铁(fe)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钌(ru)、铑(rh)、钯(pd)和铂(pt)。甚至更具体地,m选自硼(b)、铝(al)、镓(ga)、铟(in)、锗(ge)、锡(sn)、锑(sb)、钛(ti)、锆(zr)、钴(co)、镍(ni)、铜(cu)、锌(zn)、钌(ru)、铑(rh)、钯(pd)和铂(pt)。最具体地,m选自硼(b)、铝(al)、镓(ga)、铟(in)和钯(pd)。r1至r5、x、y、l、a、n、m、p分别如上述限定,或如下面进一步限定。
在另一个具体实施方案中,配合物具有式1b并且m选自镧系或锕系。更具体地,m选自镧(la)、铈(ce)、镨(pr)、钕(nd)、钐(sm)、铕(eu)、钆(gd)、铽(tb)、镝(dy)、钬(ho)、铒(er)、铥(tm)、镱(yb)、镥(lu)、锕(ac)、钍(th)、铀(u)、镎(np)、钚(pu)。甚至更具体地,m选自镧(la)、铈(ce)、镨(pr)、钕(nd)、钐(sm)、镱(yb)。r1至r5、x、y、l、a、n、m、p分别如上述限定,或如下面进一步限定。
优选地,r1是氢或具有至多5个碳原子的环状、直链或支链烷基。r1也可选自nh2、nh(ch3)、n(ch3)2、n(c2h5)2或n-吡咯烷基。在r1中碳原子数太高的情况下,得到的金属配合物不能提供需要的高挥发性。更优选地,r1是氢或具有至多4个碳原子的直链或支链烷基,或r1和r2与它们键合的氮原子一起形成吡咯烷环。更优选地,r1是氢或具有至多3个碳原子的直链或支链烷基。更优选地,r1是具有至多3个碳原子的直链烷基。甚至更优选地,r1是ch3。
优选地,r2是氢或具有至多5个碳原子的环状、直链或支链烷基。在r2中碳原子数太高的情况下,得到的金属配合物不能提供需要的高挥发性。更优选地,r2是具有至多4个碳原子的直链或支链烷基,或r2和r1与它们键合的氮原子一起形成吡咯烷环。更优选地,r2是具有至多3个碳原子的直链或支链烷基。更优选地,r2是具有至多3个碳原子的直链烷基。甚至更优选地,r2是ch3。
在特别优选的实施方案中,r1和r2都是具有至多3个碳原子的直链烷基,最优选地r1和r2都是ch3。
优选地,r3是氢或具有至多5个碳原子的环状、直链或支链烷基,nh2,nh(ch3),n(ch3)2,n(c2h5)2或n-吡咯烷基。更优选地,r3是氢、ch3、c2h5、nh2、n(ch3)2或n(c2h5)2。甚至更优选地,r3是ch3、c2h5、n(ch3)2或n(c2h5)2。最优选地,r3是ch3、c2h5或n(ch3)2。特别优选地,r3是ch3或n(ch3)2。
优选地,r4和r5相互独立地是氢或具有至多3个碳原子的直链或支链烷基,或它们与它们键合的氮原子一起形成吡咯烷环。优选地,r4和r5相互独立地是氢、ch3或c2h5。更优选地,r4和r5相互独立地是ch3或c2h5。甚至更优选地,r4和r5都是ch3。
优选地,x选自氢阴离子(h-),甲基(ch3-),乙基(c2h5-),异丙基(iso-c3h7-),叔丁基(tert-c4h9-),苯基阴离子(c6h5-),邻-、间-或对-甲苯阴离子[c6h4(ch3)]-,噻吩-2-基阴离子(c4h3s-),甲氧基(meo-),乙氧基(eto-),叔丁氧基(tert-buo-),mes-,mese-,(tert-bu)s-,(tert-bu)se-,二甲基氨基(nme2-),二乙基氨基(net2-),甲基乙基氨基(nmeet-),n-吡咯烷基[nc4h8]-,氯根(cl-)或溴根(br-)。更优选地,x选自氢阴离子(h-),甲基(ch3-),乙基(c2h5-),苯基阴离子(c6h5-),邻-、间-或对-甲苯阴离子[c6h4(ch3)]-,甲氧基(meo-),乙氧基(eto-),mes-,mese-,二甲基氨基(nme2-),二乙基氨基(net2-),甲基乙基氨基(nmeet-),氯根(cl-)或溴根(br-)。甚至更优选地,x选自氢阴离子(h-),甲基(ch3-),乙基(c2h5-),苯基阴离子(c6h5-),邻-、间-或对-甲苯阴离子[c6h4(ch3)]-,甲氧基(meo-),mes-,二甲基氨基(nme2-),二乙基氨基(net2-)或氯根(cl-)。甚至更优选地,x选自氢阴离子(h-),甲基(ch3-),乙基(c2h5-)或异丙基(iso-c3h7-)。甚至更优选地,x选自氢阴离子(h-)或甲基(ch3-)。
优选地,y选自氧基[o]2-或亚氨基[nr6]2-,其中r6是具有至多6个c原子的环状、支链或直链烷基,或者是具有至多12个c原子的取代或未取代的烷基。更优选地,y选自氧基[o]2-或亚氨基[nr6]2-,其中r6是具有至多5个c原子的支链或直链烷基或者是具有至多8个c原子的取代或未取代的烷基。甚至更优选地,y是亚氨基[nr6]2-,其中r6是具有至多4个c原子的支链或直链烷基,例如[nch3]2-、[nc2h5]2-、[n-丙基]2-、[n-异丙基]2-、[n-丁基]2-、[n-异丁基]2-、[nc2h5]2-、[ntbu]2-。甚至更优选地,y是[ntbu]2-。
优选地,l选自吡啶、二噁烷、nh3、thf、co、烷基膦或芳基膦。更优选地,l选自吡啶、nh3、co、pme3、pcy3或pph3。甚至更优选地,l选自吡啶、nh3或co。
在具体实施方案中,金属配合物具有下式2
其中
m是b、al、ga、in、zn、fe或pd;
r3是ch3或n(ch3)2;
x是氢阴离子(h-)或甲基(ch3-);
a是1或2;并且
n是0、1或2。
在另一个具体实施方案中,金属配合物具有下式3
其中
m是b、al、ga、in、zn、fe或pd,
x是氢阴离子(h-)或甲基(ch3-),
a是1或2,并且
n是0、1或2。
在这些具体的金属配合物中,至少一个n-氨基胍基配体是n-二甲基氨基-n’,n”-三甲基胍基化合物(datg)。
在另一个具体实施方案中,金属配合物具有式3,其中m是b、al、ga、in或pd,x是氢阴离子(h-)或甲基阴离子(ch3-),a是1或2,以及n是0、1或2。在这些具体的式3金属配合物中,特别地至少一个n-氨基胍基配体是n-二甲基氨基-n’,n”-三甲基胍基化合物(datg)。
在进一步具体的实施方案中,金属配合物具有下式4
其中
m是b、al、ga、in、zn、fe或pd,
x是氢阴离子(h-)或甲基(ch3-),
a是1或2,并且
n是0、1或2。
在这些可选的实施方案中,至少一个n-氨基胍基配体是n,n’-双二甲基氨基-n”-二甲基胍基化合物(bdmg)。
在其它具体的实施方案中,金属配合物具有式4,其中m是ga、zn或fe,x是氢阴离子(h-)或甲基(ch3-),a是1或2,并且n是0、1或2。在金属配合物具有式4的这些实施方案中,至少一个n-氨基胍基配体是n,n’-双二甲基氨基-n”-二甲基胍基化合物(bdmg)。在一些更具体的实施方案中,x是甲基,a和n都是1或2。
在另一个具体的实施方案中,金属配合物具有下式5
其中
m是la、ce、pr、nd,sm或yb;
r3是ch3或n(ch3)2;
x是氢阴离子(h-)或甲基(ch3-);
a是1、2或3;并且
n是0、1或2。
在另一个具体的实施方案中,金属配合物具有下式6
其中
m是ce、pr、nd、sm或yb;
x是氢阴离子(h-)或甲基(ch3-);
a是1、2或3;并且
n是0、1或2。
在甚至更具体的实施方案中,金属配合物具有式6,其中m是ce、pr、nd、sm或yb,n是0并且a是3。
本专利申请也涉及产生这些新型金属配合物的方法。
该方法包括以下步骤:
1)制备n-氨基胍配体;
2)从n-氨基胍配体制备金属配合物。
n-氨基胍配体的制备(步骤1)包括使具有通式h2n-nrxry的肼与选自硫脲和1,1-二氯甲烷胺的化合物反应。优选地,式h2n-nrxry的rx和ry相互独立地是氢或具有至多4个碳原子的直链或支链烷基。
通式h2n-nrxry的肼与选自硫脲和1,1-二氯甲烷胺的化合物反应包括在反应溶剂中混合两种组分以形成配体制备混合物。
优选地,进行具有通式h2n-nrxry的肼与选自硫脲和1,1-二氯甲烷胺的化合物的反应以使选自硫脲和1,1-二氯甲烷胺的化合物与反应溶剂混合以形成配体制备预混合物。优选地,随后将肼化合物加入到配体制备预混合物中以形成配体制备混合物。优选地,缓慢添加肼化合物到配体制备预混合物中。更优选地,滴加所述肼化合物。所述优选的顺序允许进一步提高所制备的配体的产率。配体制备预混合物和配体制备混合物可以包含进一步的化合物。
优选地,步骤1的反应溶剂选自脂肪烃、芳族溶剂、氯化溶剂、醚溶剂、醇以及它们的混合物。更优选地,步骤1)的反应溶剂选自戊烷、己烷、庚烷、苯、甲苯、二氯甲烷、氯仿、乙醚、四氢呋喃、甲醇、乙醇、异丙醇以及它们的混合物。甚至更优选地,步骤1)的反应溶剂选自二氯甲烷、氯仿和四氢呋喃。
在该方法的优选形式中,式h2n-nrxry的rx或ry相互独立地是氢或甲基。在该方法的进一步优选形式中,rx和ry都是甲基。
具有通式h2n-nrxry的肼与选自硫脲和1,1-二氯甲烷胺的化合物反应可以进一步包括活化步骤。在肼与选自1,1-二氯甲烷胺的化合物反应的情况下,优选地,不进行活化步骤。
在肼与选自硫脲的化合物反应的情况,步骤1)优选地包括活化步骤。在这些实施方案中,活化步骤优选地包括选自硫脲的化合物的硫代基的烷基化。更优选地,活化步骤包括选自硫脲的化合物的硫代基的甲基化。
在肼与选自硫脲的化合物反应包括活化步骤的实施方案中,活化步骤在配体制备预混合物和配体制备混合物形成之前进行。
优选地,活化步骤的反应时间为至少1小时,更优选地至少2小时,并且甚至更优选地至少3小时。如果反应时间太短,不能以高产率获得活化的化合物。优选地,活化步骤的反应时间不超过4天,更优选地3天,甚至更优选地2天。如果反应时间太长,沉淀的可能性增加。特别优选的是活化步骤的反应时间在6小时和36小时之间。
优选地,活化步骤的反应温度为至少-80℃,更优选地-50℃,甚至更优选地,-30℃。如果反应温度太低,反应进行不够快。优选地,反应温度不超过100℃,更优选地不超过80℃,甚至更优选地不超过60℃。如果反应温度太高,发生不期望的副反应。特别优选的是反应温度在-20℃和50℃之间。
术语“硫脲”和“1,1-二氯甲烷胺”根据本专利申请分别用于具有硫脲和1,1-二氯甲烷胺基本结构的此类化合物。此类化合物可以具有额外的取代基,例如连接至各自的基本结构的烷基。
优选地,硫脲选自二甲基硫脲、三甲基硫脲、二乙基硫脲、三乙基硫脲、二甲基乙基硫脲和二乙基甲基硫脲。更优选地,硫脲选自二甲基硫脲、三甲基硫脲和二甲基乙基硫脲。甚至更优选地,硫脲是三甲基硫脲。而且,碳酰氯亚铵盐也适合用于与肼反应。
优选地,1,1-二氯甲烷胺选自1,1-二氯甲烷甲胺、1,1-二氯甲烷二甲基胺、1,1-二氯甲烷乙胺、1,1-二氯甲烷二乙基胺和1,1-二氯甲烷甲基乙基胺。更优选地,1,1-二氯甲烷胺选自1,1-二氯甲烷甲胺、1,1-二氯甲烷二甲基胺和1,1-二氯甲烷乙胺。甚至更优选地,1,1-二氯甲烷胺是1,1-二氯甲烷二甲基胺;合适的碳酰氯亚铵盐是氯化二氯亚甲基-二甲基亚铵。该化合物具有下式
该方法的从n-氨基胍配体制备金属配合物(步骤2)包括下面反应步骤:
a)任选地使n-氨基胍配体去质子化,以形成n-氨基胍基配体;
b)提供金属配合物制备混合物,其包括n-氨基胍配体或从任选的步骤a)获得的n-氨基胍基配体、至少一种金属起始化合物和反应溶剂;
c)在任选的搅拌下温育所获得的金属配合物制备混合物;
d)蒸发反应溶剂;
e)任选地纯化留下的反应产物,优选地通过用洗涤溶剂洗涤进行纯化。
任选地,n-氨基胍配体在步骤a)中去质子化,以形成n-氨基胍基配体,根据本专利申请,其是单阴离子的。氨基胍配体的去质子化(步骤a))优选地通过用碱温育氨基胍配体进行。优选地,碱选自六甲基二硅基氨基钾(khmds)、六甲基二硅基氨基锂(lihmds)、二异丙基酰胺锂(lda)和四甲基哌啶锂(litmp)。更优选地,碱选自khmds或lihmds。甚至更优选地,碱是khmds。
优选地,步骤a)的反应时间是至少1分钟,更优选地至少2分钟并且甚至更优选地至少5分钟。如果反应时间太短,不能以高产率获得去质子化的配体。优选地,步骤a)的反应时间不超过5小时,更优选地不超过4小时,甚至更优选地不超过3小时。如果反应时间太长,沉淀的可能性增加。特别优选的是步骤a)的反应时间在10分钟和1小时之间。
优选地,步骤a)的反应温度为至少-80℃,更优选地-50℃,甚至更优选地-20℃。如果反应温度太低,反应进行不够快。优选地,反应温度不超过100℃,更优选地不超过80℃,甚至更优选地不超过60℃。如果反应温度太高,发生不期望的副反应。特别优选的是反应温度在0℃和50℃之间。
在步骤b)中,形成金属配合物制备混合物,优选地通过单步反应。单步反应是没有必要的中间分离或中间纯化步骤的反应。
金属配合物制备混合物包括n-氨基胍配体或从步骤a)获得的n-氨基胍基配体、至少一种金属起始化合物和反应溶剂。
金属配合物制备混合物的提供优选地包括将n-氨基胍配体或从步骤a)获得的n-氨基胍基配体与至少一种金属起始化合物在反应溶剂中混合,以形成金属配合物制备混合物。在方法包括步骤a)的实施方案中,金属配合物制备混合物包括n-氨基胍基配体、至少一种金属起始化合物和反应溶剂。在方法不包括步骤a)的可选实施方案中,金属配合物制备混合物包括n-氨基胍配体、至少一种金属起始化合物和反应溶剂。
可以进行n-氨基胍配体或从步骤a)获得的n-氨基胍基配体与至少一种金属起始化合物在反应溶剂中的混合,使得至少一种金属起始化合物与反应溶剂混合,以形成金属配合物制备预混合物。优选地,随后加入n-氨基胍配体或从步骤a)获得的n-氨基胍基配体至金属配合物制备预混合物,以获得金属配合物制备混合物。优选地,缓慢加入n-氨基胍配体或从步骤a)获得的n-氨基胍基配体至金属配合物制备预混合物。更优选地,滴加n-氨基胍配体或从步骤a)获得的n-氨基胍基配体。描述的优选顺序允许进一步提高金属配合物的产率。金属配合物制备预混合物和金属配合物制备混合物可包含其它化合物。
在可选的实施方案中,可以进行n-氨基胍配体或从步骤a)获得的n-氨基胍基配体与至少一种金属起始化合物在反应溶剂中混合以使n-氨基胍配体或从步骤a)获得的n-氨基胍基配体与反应溶剂混合,以形成金属配合物制备预混合物。在这样的实施方案中,随后加入至少一种金属起始化合物至金属配合物制备预混合物,以获得金属配合物制备混合物。优选地,缓慢加入至少一种金属起始化合物至金属配合物制备预混合物。更优选地,滴加至少一种金属起始化合物。
合适的金属起始化合物优选地选自金属氢化物、金属卤化物、烷基金属、芳基金属、烷氧基金属、烷硫基金属、烷砷基金属、金属腈配合物例如[pdcl2(ch3cn)2]和金属酰胺。更优选地,金属起始化合物选自金属氢化物、甲基金属、乙基金属、甲基化金属、乙基化金属、金属二甲基酰胺、金属二乙基酰胺、金属甲基乙基酰胺、金属氯化物和金属溴化物。更优选地,金属起始化合物选自金属氢化物、甲基金属、乙基金属、金属二甲基酰胺和金属氯化物。甚至更优选地,金属起始化合物选自金属氢化物、甲基金属和金属氯化物。在特别优选的实施方案中,使用一种类型的金属起始化合物。
优选地,n-氨基胍配体或n-氨基胍基配体与至少一种金属起始化合物的摩尔比是至少0.1,更优选地至少0.2并且甚至更优选地至少0.4。如果摩尔比太低,不能以高产率获得本发明的金属配合物。优选地,该比不超过值20,更优选地10并且甚至更优选地3。如果摩尔比太高,不能以高产率获得本发明的金属配合物。特别优选的是n-氨基胍配体或n-氨基胍基配体与至少一种金属起始化合物的摩尔比在0.45和2.5之间。
金属配合物制备混合物包括反应溶剂。优选地,反应溶剂是有机溶剂,选自脂肪烃、芳族溶剂、氯化溶剂、醚溶剂、醇或其混合物。更优选地,反应溶剂选自戊烷、己烷、庚烷、苯、甲苯、二氯甲烷、氯仿、乙醚、四氢呋喃、甲醇、乙醇、异丙醇或其混合物。甚至更优选地,反应溶剂选自己烷、甲苯、乙醚和四氢呋喃。
优选地,步骤c)的反应时间是至少10分钟,更优选地至少20分钟并且甚至更优选地至少30分钟。如果反应时间太短,不能以高产率获得本发明的金属配合物。优选地,步骤c)的反应时间不超过5天,更优选地不超过4天,甚至更优选地不超过3天。如果反应时间太长,沉淀的可能性增加。特别优选的是步骤c)的反应时间在45分钟和48小时之间。
优选地,步骤c)的反应温度为至少-180℃,更具体地-150℃,甚至更具体地-120℃或-78℃。如果反应温度太低,反应进行不够快。优选地,反应温度不超过200℃,更具体地不超过80℃,甚至更具体地不超过60℃。如果反应温度太高,发生不期望的副反应。因此,反应温度在-50℃和200℃之间,或-100℃和70℃之间,或-100℃和50℃之间。
反应步骤c)之后,通过蒸发分离反应溶剂(步骤d))。蒸发优选地在下述压力下进行:小于10mbar,更优选地小于1mbar,更优选地小于10-3mbar并且甚至更优选地小于10-3mbar。如果压力太高,蒸发速率太低。
留下的反应产物可以被进一步纯化(步骤e))。这优选地通过用洗涤溶剂洗涤来自步骤d)的反应产物进行。洗涤溶剂优选地选自脂肪醇和烷烃或其混合物。更优选地,洗涤溶剂选自甲醇、戊烷和己烷或其混合物。甚至更优选地,洗涤溶剂是己烷。可以进行进一步的纯化步骤。
本专利申请的方法的反应步骤1)和2)优选地在惰性气体下进行。具体而言,惰性气体选自氮气和氩气。更具体地,惰性气体是氮气。
附图说明
图1显示al(datg)me2(实施例4)晶体结构的ortep图。氮原子分别标为n1至n4。胍基团的中心碳原子标为c2。铝原子标为al1。甲基中的碳原子未标示。氢原子未显示。所有键以短棍表示。
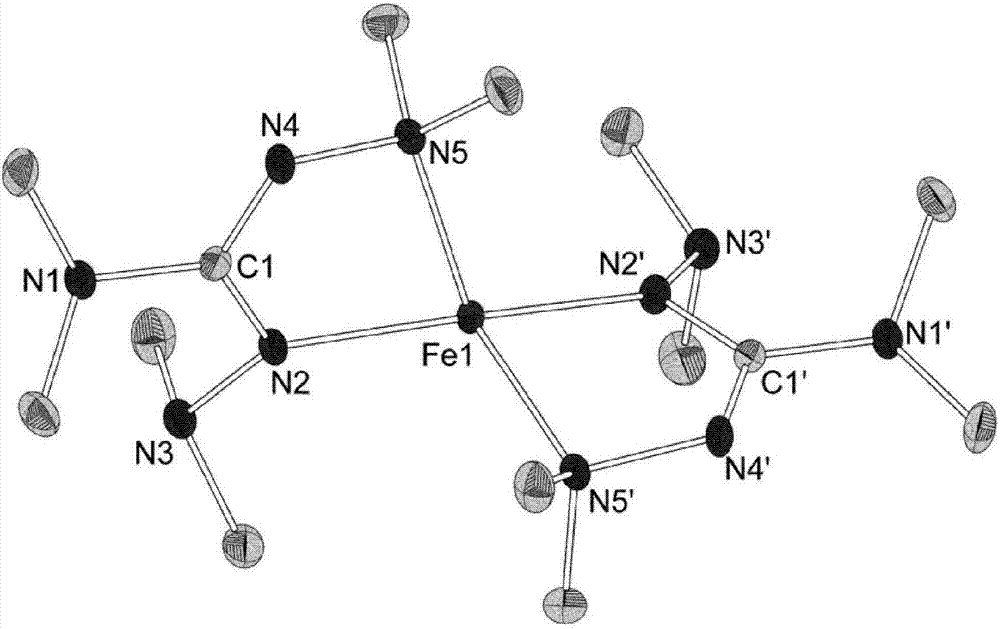
图2显示fe(bdmg)2(实施例10)晶体结构的ortep图。氮原子分别标为n1至n5或n1’至n5’。胍基团的中心碳原子标为c1和c1’。铁原子标为fe1。甲基中的碳原子未标示。氢原子未显示。所有键以短棍表示。
图3显示zn(bdmg)2(实施例11)晶体结构的ortep图。氮原子分别标为n1至n10。胍基团的中心碳原子标为c1和c8。锌原子标为zn1。甲基中的碳原子未标示。氢原子未显示。所有键以短棍表示。
图4显示yb(bdmg)3(实施例12)晶体结构的ortep图。氮原子分别标为n1至n8或n1’至n8’。胍基团的中心碳原子标为c1、c1’和c8。镱原子标为yb1。甲基中的碳原子未标示。氢原子未显示。所有键以短棍表示。
实施例
缩写
me:甲基,-ch3
dcm:二氯甲烷
dee:乙醚
mhz:兆赫兹,106s-1
mecn:乙腈
ppm:每百万之份数,nmr光谱中化学位移的单位
thf:四氢呋喃
tms:四甲基硅烷
对于nmr光谱中的多样性,缩写如下:
s:单峰
bs:宽单峰
d:双峰
ir光谱中的强度缩写如下:
w:弱
m:中等强
s:强
b:宽
一般评述
使用普通schlenk技术或惰性气体手套箱工作站(braun),在n2惰性气体条件下,进行所有合成。使用填充p4o10树脂的柱干燥使用的氮气。将真空-惰性气体管连接至旋转叶轮泵(vakuubrand)。
通过标准程序干燥使用的溶剂(w.l.f.armarego,d.d.perrin,purificationoflaboratorychemicals,4.ed.,elsevier,burlington,1996)并且存放在吸附柱中的氧化铝/分子筛
相对于作为内标的tms,以ppm表示nmr光谱的化学位移δ。残留质子或各自氘化溶剂的各自溶剂信号用于校正1h-和13c-nmr比例(1h-nmr:cdcl3:7.26ppm,c6d6:7.16ppm;13c-nmr:cdcl3:77.16ppm,c6d6:128.06ppm)。
可买到的离析物购自公司sigmaaldrich、tcieurope、alfaaesar和merck。khmds(j.
实施例1和2:制备n-氨基胍配体
实施例1:
制备n-二甲基氨基-n’,n”-三甲基胍(hdatg)
将三甲基硫脲(11.85g,100mmol,1.00当量)悬浮在0℃的20mldcm中。滴加甲基碘(mei)(6.40ml,103mmol,1.03当量)和缓慢加热溶液至室温(rt)。24小时后,在真空中浓缩此中间产物。缓慢结晶出澄清、黄色和高度粘稠的油。将mei以10mlthf溶解。在真空中除去所有其他挥发性组分。将获得的锍盐溶解在8mlmecn中。加入n,n-二甲基肼(8.5ml,112mmol,1.12当量),在40℃下搅拌溶液2小时,随后在rt下搅拌15分钟。用n2除去甲硫醇。在真空中除去mecn。用浓kohaq洗涤残留物并用dcm萃取。在真空中除去dcm,并蒸馏产物(35mbar,68℃-73℃)。获得7.64g(53.0mmol,53%)澄清产物。
1h-nmr(300mhz,c6d6):δ=5.73(s,1h,nh),2.55(s,6h,nnme2),2.46(s,6h,nme2),2.38(d,3h,j=5.7hz,nhme)。
13c-nmr(75mhz,c6d6):δ=164.9(cq),48.8(nnme2),39.8(nme2),31.3(nme)。
hr-ms(esi):m/z[m+h]+计算值:145.1448,发现值:145.1448。
ir:
实施例2:
制备n,n’-双(二甲基氨基)-n”-二甲基胍(hbdmg):
将n,n-二甲基肼(0.47ml,6.16mmol,2.0当量)和三乙胺(1.50ml,10.80mmol,3.5当量)滴加并在搅拌下加入至-10℃的5mlthf中的二氯甲基亚乙基二甲基-氯化铵(500mg,3.08mmol,1.0当量)中。1小时后,缓慢加热溶液至rt。通过过滤除去发生的沉淀并且在减压下除去挥发性组分。获得澄清油状产物(517mg,2.98mmol,97%)。
1h-nmr(300mhz,cdcl3):δ=2.80(s,6h,nme2),2.46(s,6h,nnme2)2.33(s,6h,nnme2),6.78(s,1h,nh)。
13c-nmr(75mhz,cdcl3):δ=162.1(cq),48.2(nhme2),47.1(nnme2),38.9(nme2)。
hr-ms(esi):m/z[m+h]+计算值:174.1713,发现值:174.1713。
实施例3-9:从n-氨基胍配体制备金属配合物
在下面实施例中,实施例3至5和7至9描述具有配体datg的配合物,实施例6描述具有配体bdmg的配合物。
实施例3:
制备[pd(datg)2]
将khmds(219mg,1.1mmol,2.2当量)溶于5ml甲苯,在搅拌下滴加hdatg(159mg,1.1mmol,2.2当量)。搅拌反应混合物15分钟。然后,在真空下除去溶剂和h-hmds。将获得的kdatg溶于3mlthf并缓慢滴加至3mlthf中的[pdcl2(mecn)2](200mg,0.77mmol,1.0当量)。室温下搅拌溶液30分钟,随后40℃搅拌30分钟。离心除去kcl,在真空中从橙色上清液除去溶剂。用己烷洗涤得到的橙褐色固体并在真空下干燥。获得87mg橙色固体状(0.22mmol,29%)期望产物。
1h-nmr(300mhz,c6d6):δ=2.81(s,6h,pdnme),2.56(s,12h,pdnme2),2.47(s,12h,nme2)。
13c-nmr(75mhz,c6d6):δ=160.2(cquart),53.9(pdnme2),41.9(nme2),39.4(pdnme)。
hr-ms(apci,mecn):m/z[m+h]+的计算值:393.1706,发现值:393.1701。
ir:
实施例4
制备[al(datg)me2]
将在5ml甲苯中的hdatg(415mg,2.88mmol,1.01当量)缓慢滴加至在-78℃的5ml甲苯中的三甲基铝(205mg,2.84mmol,1.00当量)中。1小时后,升温至0℃。45分钟后,在室温下搅拌12小时。随后,在真空下除去溶剂,得到淡黄色的油,其在10℃左右结晶。通过在40℃蒸发,然后浓缩,转移产物。获得无色结晶固体(135mg,674μmol,24%),其容易通过手的热熔化。
1h-nmr(300mhz,c6d6):δ=2.72(s,3h,nme),2.51(s,6h,nme2),2.19(s,6h,nme2),-0.45(s,6h,alme2)。
13c-nmr(75mhz,c6d6):δ=170.5(ch),48.7(nme2),40.6(nme2),31.8(nme),-11.2(alme2)。
ir:
熔点:没有精确测量,大约>20℃。
实施例5
制备[in(datg)me2]
将在3ml甲苯中的hdatg(290mg,2.01mmol,1.00当量)缓慢滴加至-78℃的2.5ml甲苯中的三甲基铟(321mg,2.01mmol,1.00当量)中。45分钟后升温至0℃。另外45分钟后在rt下搅拌2小时。随后,在真空下除去溶剂,获得淡品红色的油。通过在40℃蒸发然后浓缩,转移产物。获得无色液体(233mg,809μmol,40%),鉴定为[in(datg)me2]。
1h-nmr(300mhz,c6d6):δ=2.90(d,3h,j=1.1hz,nme),2.63(d,6h,j=1.5hz,nme2),2.20(s,6h,nme2),-0.09(d,6h,j=1.7hz,inme2)。
13c-nmr(75mhz,c6d6):δ=170.8(ch),50.0(nme2),41.5(nme2),35.0(nme),-8.7(inme2)。
ir:
熔点:没有精确测量,<0℃。
实施例6
制备[ga(bdmg)me2]
将-78℃下的2ml甲苯中的三甲基镓(184mg,1.60mmol,1.00当量)缓慢滴加至3ml甲苯中的hbdmg(275mg,1.60mmol,1.00当量)。1小时后升温至0℃。另外45分钟后rt下搅拌36小时。随后,在真空下除去溶剂,获得淡黄色油(167mg,0.61mmol,38%),鉴定为期望的产物。
1h-nmr(300mhz,cdcl3):δ=2.86(s,6h,nme2),2.53(s,6h,nnme2),2.48(s,6h,nnme2),-0.249(s,6h,game2)。
13c-nmr(75mhz,cdcl3):δ=166.1(cquart),48.0(nnme2),47.8(nnme2),39.7(nme2),-7.1(game2)。
实施例7
制备[b(datg)h2]
将硼氢化锂(150mg,6.8mmol,1当量)悬浮在10mlthf中。将己烷(2.2mmol,0.3当量)中的2.2ml的1mbcl3溶液缓慢滴加至-78℃的悬浮液。rt下搅拌反应混合物1小时。随后,冷却混合物至-78℃并与hdatg(1.7ml,12.7mmol,1.8当量)混合。加热至rt,搅拌过夜。使用celitetm从反应混合物除去沉淀,并在高真空下干燥澄清滤液。获得636mg(4.1mmol,60%)无色固体状产物。
1h-nmr(cdcl3,300mhz):δ/ppm=2.68(s,6h,cnme2),2.52(s,3h,nme),2.51(s,6h,bnme2)。
13c-nmr(cdcl3,75mhz):由于溶解性差,信号强度太低。
hr-ms(esi):m/z[m+h]+计算值:157.1619,发现值:157.1621。
实施例8
制备[al(datg)h2]
将lialh4(164mg,4.3mmol,2.2当量)在-78℃溶于20ml乙醚,分几份加入盐酸三甲基胺(392mg,4.1mmol,2当量)。在-78℃搅拌反应混合物1小时。随后,加热至rt并在rt下再搅拌12小时。在0℃,在恒定搅拌下滴加hdatg(0.26ml,2mmol,1当量)。加热混合物至rt并搅拌12小时。使用celitetm除去沉淀,并在高真空下干燥澄清滤液。将残留物悬浮在10ml己烷中和5ml乙醚中。过滤浑浊溶液,在高真空下除去澄清滤液的溶剂。作为产物,获得165mg(0.9mmol,48%)透明固体。
1h-nmr(cdcl3,300mhz):δ/ppm=4.02(bs,2h,alh2),2.62(s,6h,cnme2),2.47(s,9h,nme2,nme)。
13c-nmr(cdcl3,75mhz):由于溶解性差,信号强度太低。
实施例9
制备[ga(datg)h2]
将三氯化镓(0.55g,2.6mmol,1当量)溶于-78℃的5ml乙醚中并滴加至冷却至-78℃的5ml乙醚中的氢化锂(0.40g,50.3mmol,16当量)悬浮液中。在-78℃下将反应混合物再搅拌2小时。随后,加热至rt并搅拌过夜。过滤除去沉淀,并将澄清滤液在-78℃下与也冷却至-78℃的4ml乙醚中的三氯化镓(0.40g,2.3mmol,0.7当量)溶液混合。加热得到的悬浮液至0℃,随后过滤。冷却滤液至-78℃并与hdatg(0.33ml,2.5mmol,0.8当量)混合。缓慢加热得到的悬浮液至0℃并搅拌过夜。使用celitetm过滤反应混合物,并在高真空下在0℃通过蒸发浓缩澄清滤液。获得429mg白色固体状(1.9mmol,64%)产物。
1h-nmr(cdcl3,300mhz):δ/ppm=5.2(bs,2h,gah2),2.88(s,6h,cnme2),2.67(s,9h,nme2,nme)。
13c-nmr(cdcl3,75mhz):δ/ppm=169.5(cquart.),49.8(nnme),40.2(nme2),33.4(nme)。
实施例10
合成[fe(bdmg)2]
在室温下滴加hbdmg(300mg,1.73mmol,2.2当量)至甲苯(10ml)中的khmds(345mg,1.73mmol,2.2当量)中。搅拌混合物1h,在此期间形成白色沉淀,之后在减压下除去挥发性化合物。将甲苯(15ml)中的fecl2(98.8mg,0.78mmol,1.0当量)加入至甲苯(10ml)中的如此制备的kbdmg中。在90℃搅拌溶液48h,在此期间溶液变黄。在减压下除去溶剂,将深黑色残留物溶于己烷(25ml),通过硅藻土过滤,并再次在减压下除去溶剂。这导致深黄色固体形式的期望产物(136mg,0.34mmol,43%)。
1h-nmr(c6d6,300mhz):δ/ppm=-25.39(s,12h,c=nnme2),-25.18(s,12h,c-nnme2),-24.80(s,12h,cnme2)。
ir:
实施例11
合成[zn(bdmg)2]
在室温将hbdmg(2.2g,12.7mmol,2.0当量)滴加至甲苯(10ml)中的[zn(hmds)2](2.44g,6.35mmol,1.0当量)中。搅拌溶液4h,之后在减压下除去溶剂。升华白色固体产生了2.31g(5.65mmol,89%)期望产物。
1h-nmr(c6d6,300mhz):δ/ppm=2.35(s,6h,c=nnmeme),2.37(s,6h,c=nnmeme),2.46(s,12h,c-nnme2),2.96(s,12h,cnme2)。
13c-nmr(c6d6,75mhz):δ/ppm=41.1(c-nnme2,cnme2),50.2(c=nnme2),168.6(cquart)。
hr-ei-ms:calc.forc14h36n10zn:408.2416m/z,found:408.2429m/z。
ir:
实施例12
合成[yb(bdmg)3]
将hbdmg(0.11g,0.65mmol,3.2当量)加入至5mlthf中的khmds(0.13g,0.65mmol,3.2当量)溶液。搅拌混合物1h,之后在减压下除去挥发性化合物。将残留物溶于5mlthf中,并在室温下加入thf(3ml)中的[ybcl3(thf)3](0.1g,0.20mmol,1.0当量)浆液。搅拌溶液12h,之后在减压下除去溶剂,加入甲苯至残留物,并过滤掉kcl。浓缩溶液,获得黄色晶体状期望产物(62mg,0.09mmol,45%)。
1h-nmr(c6d6,300mhz):δ/ppm=2.24(s,6h,c=nn(ch3)2),2.46(s,6h,c-nn(ch3)2),2.83(s,6h,cn(ch3)2)。
- 还没有人留言评论。精彩留言会获得点赞!