末端改性可溶性多官能乙烯基芳香族共聚合物、硬化性树脂组合物及使用其的光导波管的制作方法
本发明涉及一种耐热性、相容性及韧性得到改善的新型末端改性可溶性多官能乙烯基芳香族共聚合物、及硬化性树脂组合物。尤其,本发明涉及一种含有新型末端改性可溶性多官能乙烯基芳香族共聚合物、且作为要求高度的可靠性的领域的尖端电子设备领域的基板材料有用的硬化性树脂组合物、其硬化性复合材料及层叠体,另外,本发明涉及一种透明性、光传播损耗性、耐热变色性、环境可靠性及光传输损耗性优异的光导波管形成用树脂组合物、光导波管形成用树脂膜及使用这些的光导波管。
背景技术:
:伴随近年来的信息通信量的增加,而积极地进行高频率带中的信息通信,为了更优异的电气特性,其中为了减少高频率带中的传输损耗,而正寻求一种具有低介电常数与低介电损耗正切,尤其吸水后的介电特性变化小的电气绝缘材料。进而,使用这些电气绝缘材料的印刷基板或电子零件在安装时暴露在高温的再流焊中,因此期望一种耐热性高,即显示出高玻璃化温度的材料。尤其最近因环境问题而使用熔点高的无铅的焊料,因此耐热性更高的电气绝缘材料的要求提高。针对这些要求,一直以来提出有使用具有各种化学结构的乙烯基系化合物的硬化树脂。作为此种硬化树脂,例如在专利文献1中揭示有一种可溶性多官能乙烯基芳香族共聚合物,其通过在路易斯酸催化剂及特定结构的引发剂的存在下,使二乙烯基芳香族化合物与单乙烯基芳香族化合物在有机溶媒中以20℃~100℃的温度进行聚合而获得。另外,在专利文献2中揭示有一种可溶性多官能乙烯基芳香族共聚合物的制造方法,其是在四级铵盐的存在下,利用路易斯酸催化剂及特定结构的引发剂,使含有二乙烯基芳香族化合物20摩尔%~100摩尔%而成的单量体成分以20℃~120℃的温度进行阳离子聚合,由此具有得到控制的分子量分布的可溶性多官能乙烯基芳香族共聚合物的制造方法。利用所述2个专利文献中揭示的技术所获得的可溶性多官能乙烯基芳香族共聚合物的溶剂可溶性及加工性优异,通过使用所述可溶性多官能乙烯基芳香族共聚合物,可获得玻璃化温度高且耐热性优异的硬化物。利用这些技术所获得的可溶性多官能乙烯基芳香族共聚合物因其本身具有聚合性的双键,故通过使其硬化,可提供具有高玻璃化温度的硬化物。因此,所述硬化物或可溶性多官能乙烯基芳香族共聚合物可以说是耐热性优异的聚合体或其前体。而且,所述可溶性多官能乙烯基芳香族共聚合物与其他自由基聚合性单体进行共聚合而提供硬化物,所述硬化物也成为耐热性优异的聚合体。但是,若自专利文献1及专利文献2中揭示的可溶性多官能乙烯基芳香族共聚合物与其他硬化性树脂的相容性、或硬化后的耐热变色性这一观点来看,与极性高的环氧化合物或酚树脂之间的相容性或溶解性并不充分,另外,对于高工艺温度的耐热分解性也不充分。因此,因环氧化合物或酚树脂的种类而提供不透明的组合物的情况多,难以制作环氧化合物或酚树脂与可溶性多官能乙烯基芳香族共聚合物的均匀的硬化物。其除产生调配配方设计的自由度小、及硬化物的韧性低这一缺点以外,存在因280℃~300℃附近的高热历程而产生膨胀或剥离等不良的情况。另外,在专利文献3中揭示有一种可溶性多官能乙烯基芳香族共聚合物,其是使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)进行共聚合所获得的共聚合物,在其末端基的一部分具有介隔醚键或硫醚键的链状烃基或芳香族烃基。但是,所述可溶性多官能乙烯基芳香族共聚合物的韧性不足,因此在其硬化性组合物的硬化物中,无法获得充分的力学性质,故在复合体硬化物中,存在层间剥离强度不足、可靠性下降等课题。另外,虽然记载有可使用酯化合物作为促进剂,但作为能够使用的酯化合物而具体例示者为乙酸乙酯、丙酸甲酯等不具有向可溶性多官能乙烯基芳香族共聚合物的末端导入官能基的功能的酯化合物。因此,专利文献3中揭示的可溶性多官能乙烯基芳香族共聚合物的末端基是含有源自具有醇性羟基的链状烃化合物及芳香族烃化合物、以及具有硫醇性巯基的链状烃化合物及芳香族烃化合物的介隔醚键或硫醚键的任一者的链状烃基或芳香族烃基作为末端基者。另一方面,在专利文献4及专利文献5中揭示有一种具有源自芳香族系醚化合物的末端基的多官能乙烯基芳香族共聚合物、及具有源自硫代(甲基)丙烯酸酯系化合物的末端基的可溶性多官能乙烯基芳香族共聚合物。但是,这些专利文献中揭示的可溶性多官能乙烯基芳香族共聚合物虽然韧性得到改善,但存在如下的问题点:不具有伴随近年来的信息通信量的增加的高频率带中的低介电特性,无法应用于如尖端电气·电子领域之类的要求高功能且高度的电气特性、热特性·机械特性的尖端
技术领域:
。另外,在湿热历程后,这些可溶性多官能乙烯基芳香族共聚合物与玻璃布的界面的密接性下降,因此存在无法用于要求高度的可靠性的领域的基板材料这一缺点。另一方面,在专利文献6中揭示有一种硬化性树脂组合物,其包括在两末端具有乙烯基的聚苯醚寡聚物、与具有源自包含二乙烯基芳香族化合物及乙基乙烯基芳香族化合物的单量体的结构单元的多官能乙烯基芳香族共聚合物。但是,所述使用可溶性多官能乙烯基芳香族共聚合物的硬化性树脂组合物因层间剥离强度、镀层剥离强度与湿热历程后的介电特性不足,故存在无法用作尖端电子设备领域中的基板材料这一缺点。在专利文献7中揭示有一种硬化性树脂组合物,其包括具有源自包含二乙烯基芳香族化合物及乙基乙烯基芳香族化合物的单量体的结构单元的多官能乙烯基芳香族共聚合物,与含有选自由环氧基、氰酸酯基、乙烯基、乙炔基、异氰酸酯基及羟基所组成的群组中的一种以上的官能基的热硬化性树脂。但是,所述使用可溶性多官能乙烯基芳香族共聚合物的硬化性树脂组合物因镀敷性不足,故存在无法应用于要求高度的微细化的高功能的尖端
技术领域:
这一问题点。然而,在电子元件间或配线基板间的高速·高密度信号传输中,在从前的利用电气配线的传输中,信号的相互干涉或衰减成为障碍,开始看到高速·高密度化的极限。为了打破所述极限,正在进行利用光连接电子元件间或配线基板间的技术,所谓的光互连技术的开发。作为光传输管,就加工的容易性、低成本、配线的自由度高、且能够高密度化的观点而言,聚合物光导波管受到瞩目。作为聚合物光导波管的形态,一般认为适宜的是在设想针对光电混载基板的应用的玻璃环氧树脂等硬的支撑基板上制作的刚性光导波管、或设想基板彼此的连接的不具有硬的支撑基板的柔性光导波管。进而,通过设为将柔性配线板与光导波管一体复合化而成的光电复合柔性配线板,能够进一步提升安装的自由度。就所应用的设备的使用环境或零件安装等的观点而言,对于聚合物光导波管要求透明性(低光传播损耗),并且也要求耐热性、环境可靠性。另外,就光导波管的强度及处理性的观点而言,对于强韧性的要求也正在提高。进而,关于光导波管制作工艺,需要能够简便地形成芯图案的方法,作为所述方法之一,可列举广泛用于印刷配线板制造工艺的利用曝光显影的图案形成法。作为此种材料,已知有含有(甲基)丙烯酸聚合物的光导波管材料(例如参照专利文献8~专利文献11)。专利文献8及专利文献9中所记载的光导波管材料能够利用曝光显影而形成芯图案,在波长850nm中具有透明性,且高温高湿放置试验后的光传播损耗也良好,但无耐热性的评价,例如无关于再流焊试验后的光传播损耗等的试验结果的具体的记述。另外,专利文献10中所记载的光导波管材料显示出优异的光传输损耗,且耐热性良好,但脆弱,无法满足强韧性。另外,专利文献11中所记载的光导波管材料在波长850nm中具有透明性,且强韧性优异,但无耐热性的评价,例如无关于再流焊试验后的光传播损耗等的试验结果的具体的记述,也无环境可靠性的评价,例如无关于高温高湿放置试验或温度循环试验后的光传播损耗等的试验结果的具体的记述。[现有技术文献][专利文献][专利文献1]日本专利特开2004-123873号公报[专利文献2]日本专利特开2005-213443号公报[专利文献3]日本专利特开2007-332273号公报[专利文献4]日本专利特开2010-229263号公报[专利文献5]日本专利特开2010-209279号公报[专利文献6]wo2005/73264[专利文献7]日本专利特开2006-274169号公报[专利文献8]日本专利特开2006-146162号公报[专利文献9]日本专利特开2008-33239号公报[专利文献10]日本专利特开2006-71880号公报[专利文献11]日本专利特开2007-122023号公报技术实现要素:鉴于此种
背景技术:
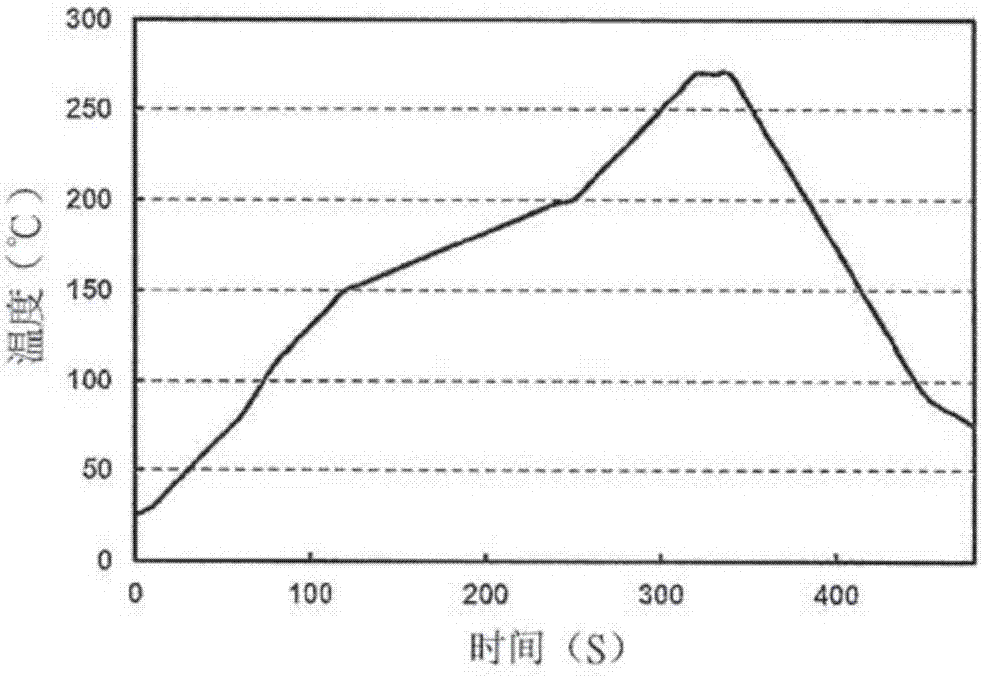
,本发明的目的在于提供一种耐热性、相容性及韧性得到改善的新型末端改性可溶性多官能乙烯基芳香族共聚合物,并且提供一种作为要求高度的可靠性的尖端电子设备领域的基板材料有用的硬化性树脂组合物、其硬化性复合材料及层叠体,另外提供一种透明性、低光传播损耗性、耐热性、环境可靠性、强韧性优异的硬化性树脂组合物,特别是可生产性及作业性良好地形成光导波管的光导波管用硬化性树脂组合物、光导波管形成用树脂膜及使用这些的光导波管。本发明人等人反复努力研究的结果,发现包含特定的末端改性可溶性多官能乙烯基芳香族共聚合物、分子中具有1个以上的不饱和基且具有特定的末端基的一种以上的乙烯基化合物、及自由基聚合引发剂的硬化性树脂组合物可解决所述课题,从而完成了本发明。即,本发明是一种末端改性可溶性多官能乙烯基芳香族共聚合物,其是包括二乙烯基芳香族化合物(a)单元及单乙烯基芳香族化合物(b)单元、与由下述式(2)及下述式(3)所表示的末端基的共聚合物,其特征在于:其具有溶剂可溶性、且具有聚合性。[化1](此处,r1表示碳数1~18的烃基或氢,r2~r3表示碳数1~18的烃基,r4表示氢或甲基)所述共聚合物优选的是二乙烯基芳香族化合物(a)单元为包含不具有乙烯基的结构单元、及具有1个乙烯基的结构单元者。所述共聚合物优选的是数量平均分子量mn为300~100,000,分子量分布(mw/mn)为100.0以下。所述共聚合物优选的是由所述式(3)所表示的末端基的导入量(c1)满足下述式(4)(c1)≧1.0(个/分子)(4),共聚合物中的源自二乙烯基芳香族化合物的结构单元的摩尔分率(a)及源自单乙烯基芳香族化合物的结构单元的摩尔分率(b)满足下述式(5)0.05≦(a)/{(a)+(b)}≦0.95(5),由所述式(2)及式(3)所表示的末端基的摩尔分率(c)满足下述式(6)0.005≦(c)/{(a)+(b)}<2.0(6),且可溶于甲苯、二甲苯、四氢呋喃、二氯乙烷或氯仿中。另外,本发明是一种末端改性可溶性多官能乙烯基芳香族共聚合物的制造方法,其制造所述末端改性可溶性多官能乙烯基芳香族共聚合物,其特征在于:在选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上的催化剂(d)的存在下,使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)、与由下述式(1)所表示的(甲基)丙烯酸酯系化合物(c)进行聚合。[化2](此处,r1~r4的含义与式(2)、式(3)相同)在所述共聚合物的制造方法中,优选的是相对于二乙烯基芳香族化合物(a)与单乙烯基芳香族化合物(b)的合计100摩尔%,使用二乙烯基芳香族化合物(a)5摩尔%~95摩尔%、单乙烯基芳香族化合物(b)95摩尔%~5摩尔%,进而相对于所有单量体100摩尔,使用由所述式(1)所表示的(甲基)丙烯酸酯系化合物(c)0.5摩尔~500摩尔,相对于(甲基)丙烯酸酯系化合物(c)1摩尔,使用催化剂(d)0.001摩尔~10摩尔,并使含有这些的聚合原料在介电常数为2.0~15.0的均匀溶媒中进行聚合。进而,本发明是一种硬化性组合物,其特征在于:包括所述末端改性可溶性多官能乙烯基芳香族共聚合物与自由基聚合引发剂。所述硬化性组合物可含有改性聚苯醚(xc)。所述硬化性组合物可含有选自由1分子中具有2个以上的环氧基与芳香族结构的环氧树脂、1分子中具有2个以上的环氧基与氰脲酸酯结构的环氧树脂、和/或1分子中具有2个以上的环氧基与脂环结构的环氧树脂所组成的群组中的一种以上的环氧树脂(xd),以及硬化剂(xe)。进而,本发明是一种使所述硬化性组合物硬化而成的硬化物、或使所述硬化性组合物成形为膜状而成的膜。进而,本发明是一种硬化性复合材料,其包括所述硬化性组合物与基材,其特征在于:以5重量%~90重量%的比例含有基材,另外,本发明是一种硬化复合材料,其特征在于:使所述硬化性复合材料硬化而获得,而且,本发明是一种层叠体,其特征在于:包括所述硬化复合材料的层与金属箔层。另外,本发明是一种带有树脂的金属箔,其特征在于:在金属箔的一面上具有由所述硬化性组合物形成的膜,或者本发明是一种电路基板材料用清漆,其是使所述硬化性组合物溶解于有机溶剂中而成。另外,本发明也为一种末端改性可溶性多官能乙烯基芳香族共聚合物,其是在选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上的催化剂(d)的存在下,使二乙烯基芳香族化合物(a)、单乙烯基芳香族化合物(b)及(甲基)丙烯酸酯系化合物(c)进行反应所获得的共聚合物,其特征在于:在所述共聚合物的末端的一部分具有分别由所述式(2)及所述式(3)表示的源自(甲基)丙烯酸酯系化合物(c)的末端基,数量平均分子量mn为300~100,000,由重量平均分子量mw与数量平均分子量mn的比所表示的分子量分布(mw/mn)为100.0以下,所述末端基的导入量(c1)满足下述式(3)(c1)≧1.0(个/分子)(3),共聚合物中的源自二乙烯基芳香族化合物的结构单元的摩尔分率(a)及源自单乙烯基芳香族化合物的结构单元的摩尔分率(b)满足下述式(4)0.05≦(a)/{(a)+(b)}≦0.95(4),所述末端基的摩尔分率(c)满足下述式(5)0.005≦(c)/{(a)+(b)}<2.0(5),且可溶于甲苯、二甲苯、四氢呋喃、二氯乙烷或氯仿中。另外,本发明也为一种末端改性可溶性多官能乙烯基芳香族共聚合物的制造方法,其是使二乙烯基芳香族化合物(a)、单乙烯基芳香族化合物(b)及(甲基)丙烯酸酯系化合物(c)进行反应来制造共聚合物的方法,其特征在于:相对于二乙烯基芳香族化合物(a)与单乙烯基芳香族化合物(b)的合计100摩尔%,使用二乙烯基芳香族化合物(a)5摩尔%~95摩尔%、单乙烯基芳香族化合物(b)95摩尔%~5摩尔%,进而相对于所有单量体100摩尔,使用由所述式(1)所表示的(甲基)丙烯酸酯系化合物(c)0.5摩尔~500摩尔,及选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上的催化剂(d),在使含有这些的聚合原料溶解于介电常数为2.0~15.0的溶媒中而成的均匀溶媒中,以20℃~120℃的温度进行聚合而获得在共聚合物的末端具有1.0(个/分子)以上的由下述式(2)~式(3)所表示的源自(甲基)丙烯酸酯系化合物(c)的末端基,且可溶于甲苯、二甲苯、四氢呋喃、二氯乙烷或氯仿中的共聚合物。进而,本发明是一种硬化性树脂组合物,其特征在于:包括(a)成分:作为具有二乙烯基芳香族化合物(a)单元及单乙烯基芳香族化合物(b)单元、与由下述式(2)及下述式(3)所表示的末端基的共聚合物,且具有聚合性以及溶剂可溶性的末端改性可溶性多官能乙烯基芳香族共聚合物;(b)成分:分子中具有1个以上的不饱和基的一种以上的乙烯基化合物;以及(c)成分:自由基聚合引发剂;且(a)成分的调配量为5wt%~94.9wt%,(b)成分的调配量为5.0wt%~85wt%,及(c)成分的调配量为0.1wt%~10wt%。(a)成分优选的是如下的具有聚合性的溶剂可溶性的末端改性可溶性多官能乙烯基芳香族共聚合物,其为具有二乙烯基芳香族化合物(a)单元及单乙烯基芳香族化合物(b)单元、与由所述式(2)及所述式(3)所表示的末端基的共聚合物,且所述二乙烯基芳香族化合物(a)单元具有由下述式(a1)所表示的结构单元、由下述式(a2)所表示的结构单元、及由下述式(a3)所表示的结构单元,所述单乙烯基芳香族化合物(b)单元具有由下述式(b)所表示的结构单元。其优选的是在选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上的催化剂(d)的存在下,使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)与由所述式(1)所表示的(甲基)丙烯酸酯化合物(c)进行聚合所获得的共聚合物,但也可为以除此以外的原料或条件进行聚合所获得的共聚合物。[化3](此处,r15、r16、r17表示碳数6~30的芳香族烃基)[化4](此处,r18表示碳数6~30的芳香族烃基。z表示碳数1~6的烷基、碳数1~6的烷氧基或氢原子)另外,本发明是一种光导波管形成用的硬化性树脂组合物,其特征在于:所述硬化性树脂组合物用于光导波管形成。在光导波管形成用的硬化性树脂组合物中,(c)成分以含有光自由基聚合引发剂为宜。另外,(b)成分以含有具有由下述通式(16)或通式(17)所表示的结构单元的乙烯基化合物、或自所述乙烯基化合物所产生的硬化型乙烯基系聚合物为宜。[化5][化6](式中,r5、r8表示氢原子或甲基,x1、x2表示可含有选自由单键、或酯键、醚键、硫酯键、硫醚键及酰胺键所组成的群组中的一种以上的键的碳数1~20的二价的有机基,r6、r9表示氢原子或碳数1~20的一价的有机基)另外,本发明是一种光导波管形成用树脂膜,其使用所述光导波管形成用的硬化性树脂组合物来形成。进而,本发明是一种光导波管,其包括使用所述光导波管形成用的硬化性树脂组合物、或所述光导波管形成用树脂膜所形成的芯部和/或包层。所述光导波管以在波长850nm的光源中的光传播损耗为0.3db/cm以下为宜。自本发明的末端改性可溶性多官能乙烯基芳香族共聚合物或含有其的材料所获得的硬化物的耐热性、相容性及韧性得到改善。另外,根据本发明的制造方法,可高效率地制造所述共聚合物。另外,通过将本发明的末端改性可溶性多官能乙烯基芳香族共聚合物用作硬化性化合物,在分子内具有分子尺寸大的自由体积、且极性基少,故可获得高度的低介电特性的硬化物,并可同时实现良好的粘接性、镀敷性与湿热历程后的介电损耗正切特性。进而,本发明的硬化性树脂组合物的透明性、耐热性、强韧性优异,能够形成高精度的厚膜,不仅作为透明材料中的硬化性树脂组合物有用,而且尤其在光导波管形成用途中有用,可制成生产性高的光导波管形成用的树脂组合物或树脂膜。另外,通过使用此种光导波管形成用的树脂组合物或树脂膜,可制成透明性、耐热性、环境可靠性、强韧性优异的光导波管。附图说明图1是说明光导波管的形态的剖面图。图2是表示回流试验中的回流炉内的温度分布的图。具体实施方式本发明的可溶性多官能乙烯基芳香族共聚合物是具有聚合性的溶剂可溶性的末端改性可溶性多官能乙烯基芳香族共聚合物,其包括含有不具有乙烯基的结构单元与具有1个乙烯基的结构单元的二乙烯基芳香族化合物(a)单元、及单乙烯基芳香族化合物(b)单元、以及由所述式(2)及式(3)所表示的末端基。所述末端改性可溶性多官能乙烯基芳香族共聚合物可为在选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上的催化剂(d)的存在下,使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)与由所述式(1)所表示的(甲基)丙烯酸酯化合物(c)进行聚合所获得的共聚合物。式中,r1表示碳数1~18的烃基或氢,r2~r3表示碳数1~18的烃基,r4表示氢或甲基。优选的r1~r3为甲基、乙基等碳数1~6的烷基。本发明的可溶性多官能乙烯基芳香族共聚合物以在催化剂(d)的存在下,使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)与(甲基)丙烯酸酯化合物(c)进行聚合所获得的共聚合物为宜,且其末端的至少一部分由源自(甲基)丙烯酸酯化合物(c)的末端基改性。所述共聚合物具有源自二乙烯基芳香族化合物(a)的结构单元及源自单乙烯基芳香族化合物(b)的结构单元。共聚合物中的源自二乙烯基芳香族化合物的结构单元的摩尔分率(a)及源自单乙烯基芳香族化合物的结构单元的摩尔分率(b)以满足(a)/{(a)+(b)}=0.05~0.95为宜。优选0.2~0.8,更优选0.3~0.7。若小于0.05,则耐热性下降,因此存在因光导波管形成工艺及电气配线形成中的热历程而无法维持良好的光导波管形状的担忧。若大于0.95,则共聚合物的分子量容易变大,另外,存在因交联密度变得过大而导致蚀刻特性恶化的担忧,且存在难以形成形状优异、光损耗小的光导波管的担忧。作为源自二乙烯基芳香族化合物(a)的结构单元,包含不具有乙烯基的结构单元与具有1个乙烯基的结构单元。可认为优选的是具有由所述式(a1)、式(a2)及式(a3)所表示的结构单元。以下,将这些结构单元称为单元(a1)~单元(a3)。具有1个乙烯基的结构单元,例如单元(a1)、单元(a3)对共聚合物赋予聚合性,使共聚合物变成多官能来作为硬化性树脂。作为不具有乙烯基的结构单元的单元(a2)赋予交联结构,增加分支度,但若交联过度进行,则硬化而变成溶剂不溶性,因此必须存在不参与交联的单元(a1)及单元(a3)。而且,相对于源自二乙烯基芳香族化合物的结构单元的摩尔分率(a)及源自单乙烯基芳香族化合物的结构单元的摩尔分率(b)的总和,共聚合物中的单元(a1)及单元(a3)的含量以10摩尔%~60摩尔%为宜,优选15摩尔%~50摩尔%,更优选20摩尔%~40摩尔%。单元(a2)的含量以5摩尔%~50摩尔%为宜,优选10摩尔%~40摩尔%。单元(a1)与单元(a3)的摩尔比以99.999:0.001~1:99的范围为宜。共聚合物中的单元(a1)与单元(a3)相比,硬化时的聚合性良好,因此优选的是以99.99:0.01~30:70的范围为宜。更优选99.99:0.01~50:50的范围。就其他观点而言,源自二乙烯基芳香族化合物的结构单元中的具有1个乙烯基的结构单元的含量以10摩尔%~90摩尔%为宜,优选20摩尔%~80摩尔%,更优选30摩尔%~70摩尔%。但是,以显示出溶剂可溶性的方式控制所述结构单元的含量或聚合度。在所述式(a1)、式(a2)及式(a3)中,r15、r16、r17表示碳数6~30的芳香族烃基,但这些源自二乙烯基芳香族化合物,因此根据其说明来理解。另外,在式(b)中,r18表示碳数6~30的芳香族烃基,z表示碳数1~6的烷基、碳数1~6的烷氧基或氢原子,但这些源自单乙烯基芳香族化合物,因此根据其说明来理解。共聚合物中的源自二乙烯基芳香族化合物(a)的结构单元的含量以5摩尔%~95摩尔%为宜,优选10摩尔~90摩尔%,更优选20摩尔~70摩尔%。若所述含量少,则伴随交联密度的下降,耐热性下降,因此当受到光导波管形成工艺等中的热历程时,难以维持良好的形状,若过多,则蚀刻特性恶化,难以形成微细结构优异的形状的光导波管等。就其他观点而言,相对于所有结构单元的合计100摩尔%,优选的是含有结构单元(a)30摩尔%~90摩尔%。结构单元(a)含有作为用以使耐热性显现的交联成分的乙烯基,另一方面,源自单乙烯基芳香族化合物的结构单元(b)不具有所述乙烯基,因此赋予成形性等。除源自二乙烯基芳香族化合物(a)的结构单元及单乙烯基芳香族化合物(b)以外,本发明的共聚合物具有源自(甲基)丙烯酸酯化合物(c)的由所述式(2)及式(3)所表示的末端基作为结构单元。若将各结构单元的存在摩尔比设为(a)、(b)、(c),则末端基的摩尔分率(c)/{(a)+(b)}为0.005以上、未满2.0,优选0.01~1.5,更优选0.05~1.0。通过以满足所述关系的方式将所述末端基导入至共聚合物的末端,可制成光损耗低、韧性高、具有优异的耐热性、与(甲基)丙烯酸酯化合物的相容性优异、成形加工性也优异的树脂组合物或制品。若末端基的摩尔分率小,则与(甲基)丙烯酸酯化合物的相容性与成形加工性下降,若末端基的摩尔分率大,则受到湿热历程后的尺寸变化变大,所述特性下降。在可溶性多官能乙烯基芳香族共聚合物每一分子中,由所述式(3)所表示的末端基的导入量(c1)平均为1.0个以上,优选2个~5个。可溶性多官能乙烯基芳香族共聚合物的mn(此处,mn是使用凝胶渗透色谱法所测定的标准聚苯乙烯换算的数量平均分子量)以300~100,000为宜,优选400~50,000,更优选500~10,000。若mn过低,则共聚合物中所含有的单官能的共聚合物的量增加,因此存在硬化物的耐热性下降的倾向,若过高,则存在容易生成凝胶、粘度变高、成形加工性下降的倾向。分子量分布(mw/mn)的值以100.0以下为宜,优选50.0以下,更优选1.5~10.0。最优选1.5~5.0。若分子量分布(mw/mn)的值过高,则存在共聚合物的加工特性下降、容易产生凝胶的倾向。可溶性多官能乙烯基芳香族共聚合物可溶于选自甲苯、二甲苯、四氢呋喃、二氯乙烷或氯仿中的溶剂中,有利的是可溶于所述溶剂的任一者中。此处,所谓可溶于溶剂中,是指在25℃下,在溶剂100g中溶解5g以上,优选10g以上。由于是可溶于溶剂中的多官能的共聚合物,因此二乙烯基芳香族化合物的乙烯基的一部分不进行交联而残存,且必须以变成适度的交联度的方式进行聚合。聚合方法将后述。可溶性多官能乙烯基芳香族共聚合物因末端由所述末端基改性,故对于(甲基)丙烯酸酯系化合物的相容性高。因此,当使含有(甲基)丙烯酸酯系化合物的硬化性树脂组合物硬化时,成为均匀硬化性或透明性优异者。继而,对可有利地制造末端改性可溶性多官能乙烯基芳香族共聚合物的制造方法进行说明。所述共聚合物通过使用路易斯酸催化剂等的催化剂(d),使二乙烯基芳香族化合物(a)及单乙烯基芳香族化合物(b)与由式(1)所表示的(甲基)丙烯酸酯化合物(c)进行聚合来制造。通常,(甲基)丙烯酸酯化合物无阳离子聚合性,因此不与乙烯基芳香族化合物进行共聚合。但是,当(甲基)丙烯酸酯化合物变成与二级醇或三级醇的酯时,在路易斯酸催化剂等酸催化剂的存在下,在二级碳或三级碳处断裂,并与二级碳阳离子或三级碳阳离子产生(甲基)丙烯酸根阴离子。然后,自二级碳阳离子或三级碳阳离子开始聚合。但是,推断所述起始反应的引发剂效率未必高,一部分成为丁烷等烷烃并被排出至系统外。另外,通过起始反应所产生的(甲基)丙烯酸根阴离子与作为活性种的末端的碳阳离子进行反应,并进行再键结,由此将(甲基)丙烯酸单元导入至停止末端。如上所述,作为源自(甲基)丙烯酸酯化合物(c)的末端基,有由所述式(2)及式(3)所表示的末端基,由式(2)所表示的末端基包含二级烷基或三级烷基,但其一部分并发变成烷烃的反应而被排出至系统外,因此可认为式(2)的末端基的量变成比式(3)的末端基(c1)的量略少的量。因此,可以说末端基的量以由末端基(c1)的量来特别规定为宜。二乙烯基芳香族化合物(a)与单乙烯基芳香族化合物(b)的使用量相对于两者的合计100摩尔%,以二乙烯基芳香族化合物(a)5摩尔%~95摩尔%,单乙烯基芳香族化合物(b)95摩尔%~5摩尔%为宜,优选的是二乙烯基芳香族化合物(a)15摩尔%~70摩尔%,单乙烯基芳香族化合物(b)85摩尔%~30摩尔%。二乙烯基芳香族化合物(a)使共聚合物进行分支而变成多官能,并且在使共聚合物热硬化时作为用以使耐热性显现的交联成分发挥重要的作用。作为二乙烯基芳香族化合物(a)的例子,可优选地使用二乙烯基苯(包含各异构体)、二乙烯基萘(包含各异构体)、二乙烯基联苯(包含各异构体),但并不限定于这些化合物。另外,这些化合物可单独使用、或将两种以上组合使用。就成形加工性及耐热变色性的观点而言,更优选二乙烯基苯(间体、对体或这些的异构体混合物)。单乙烯基芳香族化合物(b)改善共聚合物的溶剂可溶性及加工性。作为单乙烯基芳香族化合物(b)的例子,有苯乙烯、核烷基取代单乙烯基芳香族化合物、α-烷基取代单乙烯基芳香族化合物、β-烷基取代苯乙烯、烷氧基取代苯乙烯等,但并不受这些化合物限制。为了防止共聚合物的胶化,改善对于溶媒的溶解性、加工性,就成本及获得的容易性的观点而言,可特优选地使用苯乙烯、乙基乙烯基苯(包含各异构体)、乙基乙烯基联苯(包含各异构体)。就介电特性的观点而言,更优选乙基乙烯基苯(间体、对体或这些的异构体混合物)。另外,当制造共聚合物时,在无损本发明的效果的范围内,除二乙烯基芳香族化合物(a)、单乙烯基芳香族化合物(b)以外,可使用三乙烯基芳香族化合物、三乙烯基脂肪族化合物或二乙烯基脂肪族化合物及单乙烯基脂肪族化合物等其他单量体(e),并将所述单元导入至共聚合物中。作为其他单量体(e)的具体例,可列举1,3,5-三乙烯基苯、1,3,5-三乙烯基萘、1,2,4-三乙烯基环己烷、乙二醇二丙烯酸酯、丁二烯等,但并不受这些化合物限制。这些化合物可单独使用、或将两种以上组合使用。其他单量体(e)以在未满所有单量体的30摩尔%的范围内使用为宜。由此,相对于共聚合物中的结构单元的总量,将源自其他单量体成分(e)的结构单元设为未满30摩尔%的范围内。再者,当称为所有单量体时,除二乙烯基芳香族化合物(a)、单乙烯基芳香族化合物(b)等具有聚合性双键的单量体以外,包含(甲基)丙烯酸酯系化合物(c)。(甲基)丙烯酸酯系化合物(c)由所述式(1)表示,其在起始反应时与催化剂(d)之间产生断裂反应,生成包含二级碳阳离子或三级碳阳离子的聚合活性种,并且通过所述起始反应所产生的(甲基)丙烯酸根阴离子作为链转移剂,与作为聚合活性种的末端的碳阳离子进行再键结,由此能够对共聚合物的末端赋予韧性、低光传播损耗性、加工性等功能。自(甲基)丙烯酸酯系化合物(c)所产生的末端基由式(2)、及式(3)表示,可认为分别键结于共聚合物的起始末端及停止末端。如上所述,(甲基)丙烯酸酯系化合物(c)为单量体的一种,但也为聚合添加剂。其在共聚合物中,通过链转移反应而赋予所述末端基(结构单元之一),因此也为链转移剂。作为(甲基)丙烯酸酯系化合物(c),就反应性、获得的容易性、硬化物的耐热性的观点而言,可优选地使用甲基丙烯酸叔丁酯、甲基丙烯酸仲丁酯、丙烯酸叔丁酯、或丙烯酸仲丁酯。就反应速度的观点而言,可更优选地使用甲基丙烯酸叔丁酯、或丙烯酸叔丁酯。相对于二乙烯基芳香族化合物(a)、及单乙烯基芳香族化合物(b)的合计100摩尔,(甲基)丙烯酸酯系化合物(c)的使用量以0.5摩尔~300摩尔为宜,优选1摩尔~200摩尔,更优选10摩尔~150摩尔。若其使用量过少,则不仅所述末端基的导入量减少,韧性等功能下降,而且分子量及分子量分布增大,成形加工性恶化。另外,若其使用量过多,则除聚合速度显著下降,生产性下降以外,折射率下降。在聚合反应时,(甲基)丙烯酸酯系化合物(c)与催化剂(d)进行反应,使聚合反应开始,并且形成所述末端基后停止成长。以使源自(甲基)丙烯酸酯系化合物(c)的末端基的导入量变成共聚合物的说明中所设定的范围的方式选定其使用量及反应条件。聚合反应以使用二乙烯基芳香族化合物(a)、单乙烯基芳香族化合物(b)及(甲基)丙烯酸酯系化合物(c)与催化剂(d),在使含有这些的聚合原料类溶解于介电常数为2.0~15.0的溶媒中而成的均匀溶媒中,以20℃~120℃的温度进行阳离子共聚合而获得末端改性共聚合物为宜。作为催化剂(d),使用选自由路易斯酸催化剂、无机强酸及有机磺酸所组成的群组中的一种以上。作为路易斯酸催化剂,只要是包含金属离子(酸)与配体(碱)、且可接收电子对的化合物,则可无特别限制地使用。路易斯酸催化剂之中,就所获得的共聚合物的耐热分解性的观点而言,特优选b、al、ga、in、si、ge、sn、pb、sb、bi、ti、w、zn、fe及v等的二价~六价的金属氟化物。另外,作为无机强酸,可列举硫酸、盐酸、磷酸等。作为有机磺酸的具体例,可列举苯磺酸、对甲苯磺酸等。这些催化剂可单独使用、或将两种以上组合使用。就所获得的共聚合物的分子量及分子量分布的控制及聚合活性的观点而言,可最优选地使用三氟化硼的醚(二乙基醚、二甲基醚等)络合物。催化剂(d)以相对于(甲基)丙烯酸酯系化合物(c)1摩尔,在0.001摩尔~10摩尔的范围内使用为宜,更优选0.001摩尔~1摩尔。若超过10摩尔,则聚合速度变得过大,因此不仅难以控制分子量分布,而且源自化合物(c)的末端基的导入量减少。在制造末端改性可溶性多官能乙烯基芳香族共聚合物时,可根据期望而将选自乙酸乙酯、乙酸正丙酯、及乙酸正丁酯等自一级醇与羧酸((甲基)丙烯酸除外)所获得的酯化合物,或如甲基乙基酮、及甲基异丁基酮之类的碳数1~30的二烷基酮中的一种以上的化合物用作促进剂(f)。相对于二乙烯基芳香族化合物(a)、及单乙烯基芳香族化合物(b)的合计100摩尔,促进剂(f)的使用量未满300摩尔,优选未满200摩尔,更优选未满150摩尔。在聚合反应时,促进剂与活性种或催化剂(d)相互作用,对于提高反应的选择性、控制分子量而言有效,但若过度使用,则聚合速度显著下降,共聚合物的产率下降。所述聚合反应以在作为溶解所生成的末端改性可溶性多官能乙烯基芳香族共聚合物的溶媒的介电常数为2~15的一种以上的有机溶媒中进行为宜。作为有机溶媒,只要是本质上不阻碍阳离子聚合,且溶解催化剂、聚合添加剂、促进剂、单量体及多官能乙烯基芳香族共聚合物,形成均匀溶液的化合物,且介电常数为2~15的范围内,则并无特别限制,可单独使用、或将两种以上组合使用。若溶媒的介电常数低,则分子量分布变宽广,若溶媒的介电常数大,则聚合速度下降。作为有机溶媒,就聚合活性、溶解性的平衡的观点而言,优选甲苯、二甲苯、正己烷、环己烷、甲基环己烷及乙基环己烷。另外,考虑到所获得的聚合溶液的粘度或除热的容易性,以在聚合结束时聚合溶液中的共聚合物的浓度变成1wt%~90wt%,优选变成10wt%~80wt%,特优选变成20wt%~70wt%的方式决定溶媒的使用量。当所述浓度未满1wt%时,因聚合效率低而招致成本的增大,若超过90wt%,则分子量及分子量分布增大,招致成形加工性的下降。聚合反应温度以20℃~120℃为宜,优选40℃~100℃。若聚合温度过高,则反应的选择性下降,因此产生分子量分布的增大或凝胶的产生等问题点,若过低,则催化剂活性显著下降,必须添加大量的催化剂。聚合反应停止后,回收共聚合物的方法并无特别限定,例如只要使用汽提法、利用不良溶媒的析出等通常所使用的方法即可。根据本发明的制造方法,可有利地获得本发明的末端改性可溶性多官能乙烯基芳香族共聚合物。继而,对本发明的硬化性组合物说明第一形态。所述第一形态的硬化性组合物作为尖端电子设备领域的基板材料,例如电气绝缘材料或面向层叠体的材料有用。第一形态的硬化性组合物含有末端改性可溶性多官能乙烯基芳香族共聚合物(xa)与自由基聚合引发剂(也称为自由基聚合催化剂)(xb)。作为自由基聚合引发剂(xb),例如本发明的树脂组合物如后文所述通过加热等手段来产生交联反应而硬化,但为了降低此时的反应温度、或促进不饱和基的交联反应,可含有自由基聚合引发剂(xb)来使用。以(xa)成分与(xb)成分的和为基准,用于所述目的的自由基聚合引发剂的量为0.01重量%~10重量%,优选0.1重量%~8重量%。自由基聚合引发剂为自由基聚合催化剂,因此以下由自由基聚合引发剂来代表。自由基聚合引发剂可使用公知的物质。若列举具有代表性的例子,有苯甲酰基过氧化物、枯烯氢过氧化物、2,5-二甲基己烷-2,5-二氢过氧化物、2,5-二甲基-2,5-二(过氧化叔丁基)己炔-3、二-叔丁基过氧化物、叔丁基枯基过氧化物、α,α'-双(过氧化叔丁基-间异丙基)苯、2,5-二甲基-2,5-二(过氧化叔丁基)己烷、二枯基过氧化物、过氧化间苯二甲酸二-叔丁酯、过氧化苯甲酸叔丁酯、2,2-双(过氧化叔丁基)丁烷、2,2-双(过氧化叔丁基)辛烷、2,5-二甲基-2,5-二(过氧化苯甲酰基)己烷、二(三甲基硅烷基)过氧化物、三甲基硅烷基三苯基硅烷基过氧化物等过氧化物,但并不限定于这些过氧化物。另外,2,3-二甲基-2,3-二苯基丁烷虽然并非过氧化物,但也可用作自由基聚合引发剂(或聚合催化剂)。但是,用于本树脂组合物的硬化的催化剂、自由基聚合引发剂并不限定于这些例子。若相对于末端改性可溶性多官能乙烯基芳香族共聚合物(xa),自由基聚合引发剂的调配量为0.01重量份~10重量份的范围,则不会阻碍硬化反应而良好地进行反应。另外,在含有末端改性可溶性多官能乙烯基芳香族共聚合物(xa)的硬化性组合物中,视需要可调配能够与本发明的末端改性可溶性多官能乙烯基芳香族共聚合物(xa)进行共聚合的其他聚合性单体来进行硬化。能够进行共聚合的聚合性单体可使用公知的物质。若列举具有代表性的例子,则可列举:苯乙烯、苯乙烯二聚体、α-甲基苯乙烯、α-甲基苯乙烯二聚体、二乙烯基苯、乙烯基甲苯、叔丁基苯乙烯、氯苯乙烯、二溴苯乙烯、乙烯基萘、乙烯基联苯、苊、二乙烯基苄基醚、烯丙基苯基醚等。另外,在含有本发明的末端改性可溶性多官能乙烯基芳香族共聚合物(xa)的硬化性组合物中,也能够调配已知的热硬化性树脂,例如乙烯酯树脂、聚乙烯基苄基树脂、不饱和聚酯树脂、硬化型乙烯基树脂、改性聚苯醚树脂、顺丁烯二酰亚胺树脂、环氧树脂、聚氰酸酯树脂、酚树脂等,或已知的热塑性树脂,例如聚苯乙烯、聚苯醚、聚醚酰亚胺、聚醚砜、聚苯硫醚(polyphenylenesulfide,pps)树脂、聚环戊二烯树脂、聚环烯烃树脂等,或已知的热塑性弹性体,例如苯乙烯-乙烯-丙烯共聚合物、苯乙烯-乙烯-丁烯共聚合物、苯乙烯-丁二烯共聚合物、苯乙烯-异戊二烯共聚合物、氢化苯乙烯-丁二烯共聚合物、氢化苯乙烯-异戊二烯共聚合物等,或橡胶类,例如聚丁二烯、聚异戊二烯。本发明的硬化性组合物也可在含有末端改性可溶性多官能乙烯基芳香族共聚合物(xa)、及自由基聚合引发剂(xb)的硬化性组合物中,含有由下述式(7)所表示的改性聚苯醚(xc),特别是包含末端具有至少一个聚合性的不饱和双键的基,例如酚性羟基、乙烯基、(甲基)丙烯酸基的改性聚苯醚(xc)。本发明的末端改性可溶性多官能乙烯基芳香族共聚合物(xa)与所述改性聚苯醚(xc)具有良好的相容性,克服伴随相容性的下降的可靠性的下降这一问题,以任意的调配显示出高度的低介电特性与韧性、以及成形性与层间剥离强度等特性经改良的特性。[化7]式(7)中,m表示1或2,l表示由下述式(8)所表示的聚苯醚链。m表示氢原子、由下述式(9)所表示的基,当m为1时,m不为氢原子,当m为2时,2个m的至少任一者不为氢原子。当m为1时,t表示氢原子,当m为2时,t表示亚烷基、由下述式(10)或式(11)所表示的基。[化8]式(8)中,n表示50以下的正的整数,r5、r6、r7、及r8分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。[化9]式(9)中,x为碳数1以上的有机基,有时也包含氧原子。y为乙烯基。j表示0或1的整数。[化10]式(10)中,r10、r11、r12、及r13分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。[化11]式(11)中,r14、r15、r16、r17、r18、r19、r20、及r21分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。f为包含碳数0的情况的碳数20以下的直链状、分支状或环状的烃基。另外,由式(7)所表示的改性聚苯醚为式(7)中的m表示1或2者。即,具体而言,由式(7)所表示的改性聚苯醚为由t-l-m、或ml-t-l-m所表示的改性聚苯醚。另外,l表示由式(8)所表示的聚苯醚链。在式(8)中,n表示50以下的正的整数。另外,r5、r6、r7、及r8分别独立。即,r5、r6、r7、及r8分别可为相同的基,也可为不同的基。另外,r5、r6、r7、及r8表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。其中,优选氢原子及烷基。另外,m表示氢原子、由式(9)所表示的基。另外,m在m为1的情况下,即在改性聚苯醚为t-l-m的情况下不为氢原子,表示由式(9)所表示的基。另外,m在m为2的情况下,即在改性聚苯醚为m-l-t-l-m的情况下,2个m的至少任一者不为氢原子,就硬化物的耐热性与韧性这一理由而言,优选的是2个m均为由式(9)所表示的基。另外,t在m为1的情况下,即在改性聚苯醚为t-l-m的情况下表示氢原子。由t-l-m所表示的改性聚苯醚为由h-l-m所表示的改性聚苯醚。另外,t在m为2的情况下,即在改性聚苯醚为m-l-t-l-m的情况下,表示亚烷基、由式(10)所表示的基、或由式(11)所表示的基。其中,就硬化物的韧性与改性聚苯醚的可溶性这一理由而言,优选的是m为2,t为亚烷基,更优选m为2,t为2,2-亚丙基。另外,式(10)中,r10、r11、r12、及r13分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。r10、r11、r12、及r13分别可为相同的基,也可为不同的基。作为在r5~r21中所列举的各官能基,具体而言,可列举如下的基。烷基并无特别限定,例如优选碳数1~18的烷基,更优选碳数1~10的烷基。具体而言,例如可列举:甲基、乙基、丙基、己基、及癸基等。另外,烯基并无特别限定,例如优选碳数2~18的烯基,更优选碳数2~10的烯基。具体而言,例如可列举:乙烯基、烯丙基、及3-丁烯基等。另外,炔基并无特别限定,例如优选碳数2~18的炔基,更优选碳数2~10的炔基。具体而言,例如可列举:乙炔基、及丙-2-炔-1-基(炔丙基)等。另外,烷基羰基只要是经烷基取代的羰基,则并无特别限定,例如优选碳数2~18的烷基羰基,更优选碳数2~10的烷基羰基。具体而言,例如可列举:乙酰基、丙酰基、丁酰基、异丁酰基、三甲基乙酰基、己酰基、辛酰基、及环己基羰基等。另外,烯基羰基只要是经烯基取代的羰基,则并无特别限定,例如优选碳数3~18的烯基羰基,更优选碳数3~10的烯基羰基。具体而言,例如可列举:丙烯酰基、甲基丙烯酰基、及丁烯酰基等。另外,炔基羰基只要是经炔基取代的羰基,则并无特别限定,例如优选碳数3~18的炔基羰基,更优选碳数3~10的炔基羰基。具体而言,例如可列举:炔丙酰基等。另外,改性聚苯醚的数量平均分子量并无特别限定,但优选800~7000,更优选1000~5000。最优选1000~3000。另外,如上所述,n为50以下的正的整数,但优选的是如改性聚苯醚的数量平均分子量变成此种范围内的数值。具体而言,优选1~50。再者,此处,数量平均分子量只要是利用一般的分子量测定方法所测定者即可,具体而言,可列举使用凝胶渗透色谱法(gelpermeationchromatography,gpc)所测定的值等。若改性聚苯醚的数量平均分子量为此种范围内,则所获得的硬化性组合物的硬化物的韧性与成形性变得更高。其原因在于:若改性聚苯醚的数量平均分子量为此种范围内,则为分子量比较低者,因此一面维持韧性,一面改良流动性。通常的聚苯醚在使用此种低分子量者的情况下,存在硬化物的耐热性与韧性下降的倾向。但是,本实施方式中所使用的改性聚苯醚因在末端具有聚合性的不饱和双键,故通过与乙烯基系的热交联型硬化性树脂一同进行硬化,改性聚苯醚与热交联型硬化性树脂的交联适宜地进行,可获得硬化物的耐热性与韧性足够高者。由此,所获得的硬化性组合物的硬化物可获得耐热性及韧性均优异者。就提升异种材料间的粘接可靠性这一理由而言,本发明的硬化性组合物为如下的硬化性组合物也为适宜的实施方式,所述硬化性组合物的特征在于:在包含末端改性可溶性多官能乙烯基芳香族共聚合物(xa)、及自由基聚合引发剂(xb)的硬化性组合物中含有环氧树脂(xd)及硬化剂(xe)。作为(xd)成分的环氧树脂,并无特别限制,作为环氧树脂,优选使用选自由1分子中具有2个以上的环氧基与芳香族结构的环氧树脂、1分子中具有2个以上的环氧基与氰脲酸酯结构的环氧树脂、和/或1分子中具有2个以上的环氧基与脂环结构的环氧树脂所组成的群组中的一种以上的环氧树脂。作为(xd)成分,更优选选自由双酚a型环氧树脂、双酚f型环氧树脂、双酚s型环氧树脂、烷基苯酚酚醛清漆型环氧树脂、亚二甲苯基改性苯酚酚醛清漆型环氧树脂、亚二甲苯基改性烷基苯酚酚醛清漆型环氧树脂、联苯型环氧树脂、二环戊二烯型环氧树脂、萘型环氧树脂、三缩水甘油基异氰脲酸酯、环己烷型环氧树脂及金刚烷型环氧树脂所组成的群组中的一种以上的环氧树脂。作为用作(xd)成分的双酚f型环氧树脂,例如可列举:将4,4'-亚甲基双(2,6-二甲基苯酚)的二缩水甘油醚作为主成分的环氧树脂、将4,4'-亚甲基双(2,3,6-三甲基苯酚)的二缩水甘油醚作为主成分的环氧树脂、将4,4'-亚甲基双酚的二缩水甘油醚作为主成分的环氧树脂。其中,优选的是将4,4'-亚甲基双(2,6-二甲基苯酚)的二缩水甘油醚作为主成分的环氧树脂。作为所述双酚f型环氧树脂,能够以作为市售品的新日铁住金化学股份有限公司制造的商品名yslv-80xy的形态获得。作为联苯型环氧树脂,可列举:4,4'-二缩水甘油基联苯、及4,4'-二缩水甘油基-3,3',5,5'-四甲基联苯等环氧树脂。作为所述联苯型环氧树脂,能够以作为市售品的三菱化学股份有限公司制造的商品名yx-4000、yl-6121h的形态获得。作为二环戊二烯型环氧树脂,可列举:二氧化二环戊二烯、及具有二环戊二烯骨架的苯酚酚醛清漆环氧单体等。作为萘型环氧树脂,可列举:1,2-二缩水甘油基萘、1,5-二缩水甘油基萘、1,6-二缩水甘油基萘、1,7-二缩水甘油基萘、2,7-二缩水甘油基萘、三缩水甘油基萘、及1,2,5,6-四缩水甘油基萘、萘酚·芳烷基型环氧树脂、萘骨架改性甲酚酚醛清漆型环氧树脂、甲氧基萘改性甲酚酚醛清漆型环氧树脂、亚萘基醚型环氧树脂、甲氧基萘二亚甲基型环氧树脂等改性萘型环氧树脂等。另外,作为金刚烷型环氧树脂,可列举:1-(2,4-二缩水甘油氧基苯基)金刚烷、1-(2,3,4-三缩水甘油氧基苯基)金刚烷、1,3-双(2,4-二缩水甘油氧基苯基)金刚烷、1,3-双(2,3,4-三缩水甘油氧基苯基)金刚烷、2,2-双(2,4-二缩水甘油氧基苯基)金刚烷、1-(2,3,4-三羟基苯基)金刚烷、1,3-双(2,4-二羟基苯基)金刚烷、1,3-双(2,3,4-三羟基苯基)金刚烷、及2,2-双(2,4-二羟基苯基)金刚烷等。所述环氧树脂之中,就与(xa)成分的相容性、介电特性、及成形品的翘曲小的观点而言,可适宜地使用:双酚f型环氧树脂、烷基苯酚酚醛清漆型环氧树脂、亚二甲苯基改性苯酚酚醛清漆型环氧树脂、亚二甲苯基改性烷基苯酚酚醛清漆型环氧树脂、联苯型环氧树脂、二环戊二烯型环氧树脂、萘型环氧树脂、三缩水甘油基异氰脲酸酯、环己烷型环氧树脂、金刚烷型环氧树脂。用作(xd)成分的环氧树脂的重量平均分子量(mw)优选的是未满1万。更优选的mw为600以下,进而更优选200以上、550以下。当mw未满200时,存在所述成分的挥发性变高,浇铸膜·片的处理性变差的倾向。另一方面,若mw超过1万,则存在浇铸膜·片容易变硬且变脆、浇铸膜·片的硬化物的粘接性下降的倾向。相对于(xa)成分100重量份,(xd)成分的含量优选的是下限为5重量份、且上限为100重量份。更优选相对于(xa)成分100重量份,(xd)成分的含量的更优选的下限为10重量份。另一方面,更优选的上限为80重量份,进而更优选的上限为60重量份。若(xd)成分的含量满足所述优选的下限,则可进一步提高浇铸膜·片的硬化物的粘接性。若(xd)成分的含量满足所述优选的上限,则未硬化状态下的浇铸膜·片的操作性进一步变高,与玻璃布的密接性得到改良,可靠性变高。(xe)成分的硬化剂优选的是酚树脂、或者具有芳香族骨架或脂环式骨架的酸酐、所述酸酐的氢化物、所述酸酐的改性物、羟基末端聚苯醚寡聚物、及活性酯化合物。通过使用这些优选的硬化剂,可获得成为耐热性、耐湿性及介电特性的平衡优异的硬化物的硬化性组合物。用作(xe)成分的硬化剂的酚树脂并无特别限定。作为所述酚树脂的具体例,可列举:苯酚酚醛清漆、邻甲酚酚醛清漆、对甲酚酚醛清漆、叔丁基苯酚酚醛清漆、二环戊二烯甲酚、聚对乙烯基苯酚、双酚a型酚醛清漆、苯酚芳烷基树脂、萘酚芳烷基树脂、联苯型苯酚酚醛清漆树脂、联苯型萘酚酚醛清漆树脂、十氢萘改性酚醛清漆、聚(二-邻羟基苯基)甲烷、聚(二-间羟基苯基)甲烷、及聚(二-对羟基苯基)甲烷等。其中,优选具有三聚氰胺骨架的酚树脂、具有三嗪骨架的酚树脂、或具有烯丙基的酚树脂,其原因在于:可进一步提高绝缘片的柔软性及阻燃性。作为所述酚树脂的市售品,可列举:meh-8005、meh-8010及neh-8015(以上均为明和化成公司制造),ylh903(日本环氧树脂(japanepoxyresins)公司制造),la-7052、la-7054、la-7751、la-1356及la-3018-50p(以上均为迪爱生(dic)公司制造),以及ps6313及ps6492(群荣化学公司制造)等。关于用作(xe)成分的硬化剂的具有芳香族骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物,结构也无特别限定。作为具有芳香族骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物,例如可列举:苯乙烯/顺丁烯二酸酐共聚合物、二苯甲酮四羧酸酐、均苯四甲酸酐、偏苯三甲酸酐、4,4'-氧基二邻苯二甲酸酐、苯基乙炔基邻苯二甲酸酐、甘油双(脱水偏苯三酸酯)单乙酸酯、乙二醇双(脱水偏苯三酸酯)、甲基四氢邻苯二甲酸酐、甲基六氢邻苯二甲酸酐、及三烷基四氢邻苯二甲酸酐、甲基纳迪克酸酐、三烷基四氢邻苯二甲酸酐、或者具有二环戊二烯骨架的酸酐或所述酸酐的改性物等。作为所述具有芳香族骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物的市售品,可列举:sma树脂(resin)ef30、sma树脂(resin)ef40、sma树脂(resin)ef60及sma树脂(resin)ef80(以上均为日本沙多玛(sartomer·japan)公司制造),odpa-m及pepa(以上均为玛耐科(manac)公司制造),利卡西(rikacid)mta-10、利卡西(rikacid)mta-15、利卡西(rikacid)tmta、利卡西(rikacid)tmeg-100、利卡西(rikacid)tmeg-200、利卡西(rikacid)tmeg-300、利卡西(rikacid)tmeg-500、利卡西(rikacid)tmeg-s、利卡西(rikacid)th、利卡西(rikacid)ht-1a、利卡西(rikacid)hh、利卡西(rikacid)mh-700、利卡西(rikacid)mt-500、利卡西(rikacid)dsda及利卡西(rikacid)tda-100(以上均为新日本理化公司制造),以及艾比克隆(epiclon)b4400、艾比克隆(epiclon)b650、及艾比克隆(epiclon)b570(以上均为迪爱生公司制造)等。所述具有脂环式骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物优选的是具有多脂环式骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物,或者通过萜烯系化合物与顺丁烯二酸酐的加成反应而获得的具有脂环式骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物。在此情况下,可进一步提高绝缘片的柔软性、耐湿性或粘接性。另外,作为所述具有脂环式骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物,也可列举:甲基纳迪克酸酐、具有二环戊二烯骨架的酸酐或所述酸酐的改性物等。作为所述具有脂环式骨架的酸酐、所述酸酐的氢化物或所述酸酐的改性物的市售品,可列举:利卡西(rikacid)hna及利卡西(rikacid)hna-100(以上均为新日本理化公司制造),以及艾比固(epicure)yh306、艾比固(epicure)yh307、艾比固(epicure)yh308h及艾比固(epicure)yh309(以上均为日本环氧树脂公司制造)等。作为(xe)成分,也可使用羟基末端聚苯醚寡聚物。可列举由下述式(12)所表示的羟基末端聚苯醚寡聚物。[化12]式(12)中,p表示1或2,e表示由下述式(13)所表示的聚苯醚链,g表示氢原子,p表示1或2的整数。当p为1时,v表示氢原子,当p为2时,v表示亚烷基、由下述式(14)或式(15)所表示的基。[化13]式(13)中,q表示50以下的正的整数,r22、r23、r24、及r25分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。[化14](式(14)中,r26、r27、r28、及r29分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基)[化15]式(15)中,r30、r31、r32、r33、r34、r35、r36、及r37分别独立地表示氢原子、烷基、烯基、炔基、甲酰基、烷基羰基、烯基羰基、或炔基羰基。f为包含碳数0的情况的碳数20以下的直链状、分支状或环状的烃基。作为(xe)成分,也可使用活性酯化合物。只要是具有活性酯基者即可,但在本发明中,优选的是分子内具有至少两个活性酯基的化合物。作为用作(xe)成分的活性酯化合物,就耐热性等的观点而言,优选的是自使羧酸化合物和/或硫代羧酸化合物与羟基化合物和/或硫醇化合物进行反应而成者所获得的活性酯化合物,更优选自使羧酸化合物与选自由酚化合物、萘酚化合物及硫醇化合物所组成的群组中的一种或两种以上进行反应而成者所获得的活性酯化合物,在本发明中,特优选自使羧酸化合物与具有酚性羟基的芳香族化合物进行反应而成者获得、且分子内具有至少两个活性酯基的芳香族化合物。用作(xe)成分的活性酯化合物可为直链状或多分支状,若例示活性酯化合物源自分子内具有至少两种羧酸的化合物的情况,则当此种分子内具有至少两种羧酸的化合物含有脂肪族链时,可提高与环氧树脂的相容性,另外,当具有芳香族环时,可提高耐热性。作为用以形成活性酯化合物的羧酸化合物的具体例,可列举:苯甲酸、乙酸、丁二酸、顺丁烯二酸、衣康酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、均苯四甲酸等。这些之中,就耐热性的观点而言,优选丁二酸、顺丁烯二酸、衣康酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸,更优选邻苯二甲酸、间苯二甲酸、对苯二甲酸,进而更优选间苯二甲酸、对苯二甲酸。作为用以形成活性酯化合物的硫代羧酸化合物的具体例,可列举:硫代乙酸、硫代苯甲酸等。作为用以形成活性酯化合物的酚化合物及萘酚化合物的具体例,可列举:对苯二酚、间苯二酚、双酚a、双酚f、双酚s、酚酞啉、甲基化双酚a、甲基化双酚f、甲基化双酚s、苯酚、邻甲酚、间甲酚、对甲酚、儿茶酚、α-萘酚、β-萘酚、1,5-二羟基萘、1,6-二羟基萘、2,6-二羟基萘、二羟基二苯甲酮、三羟基二苯甲酮、四羟基二苯甲酮、均苯三酚、苯三醇、二环戊二烯基二苯酚、苯酚酚醛清漆等。这些之中,就耐热性、溶解性的观点而言,优选1,5-二羟基萘、1,6-二羟基萘、2,6-二羟基萘、二羟基二苯甲酮、三羟基二苯甲酮、四羟基二苯甲酮、二环戊二烯基二苯酚、苯酚酚醛清漆,更优选二羟基二苯甲酮、三羟基二苯甲酮、四羟基二苯甲酮、二环戊二烯基二苯酚、苯酚酚醛清漆,进而更优选二环戊二烯基二苯酚、苯酚酚醛清漆。作为用以形成活性酯化合物的硫醇化合物的具体例,可列举:苯二硫醇、三嗪二硫醇等。作为活性酯化合物,例如可使用:日本专利特开2002-12650号公报及日本专利特开2004-277460号公报中揭示的活性酯化合物、或市售的活性酯化合物。作为市售的活性酯化合物,例如可列举:商品名“exb9451、exb9460、exb9460s、hpc-8000-65t”(以上,迪爱生公司制造)、商品名“dc808”(日本环氧树脂公司制造)、商品名“ylh1026”(日本环氧树脂公司制造)等。活性酯化合物的制造方法并无特别限定,可利用公知的方法来制造,例如可通过羧酸化合物和/或硫代羧酸化合物与羟基化合物和/或硫醇化合物的缩合反应而获得。相对于环氧树脂(xd)100重量份,本发明的硬化性组合物中的活性酯化合物(xe)的调配量优选20重量份~120重量份,更优选40重量份~100重量份,进而更优选50重量份~90重量份的范围。通过将活性酯化合物(xe)的调配量设为所述范围,可提升作为硬化物的介电特性、及耐热性、线膨胀系数。作为用作(xe)成分的硬化剂,就与本发明的(xa)成分的相容性及耐湿性、粘接性的观点而言,优选的是自邻甲酚酚醛清漆、对甲酚酚醛清漆、叔丁基苯酚酚醛清漆、二环戊二烯甲酚、聚对乙烯基苯酚、亚二甲苯基改性酚醛清漆、聚(二-邻羟基苯基)甲烷、聚(二-间羟基苯基)甲烷、聚(二-对羟基苯基)甲烷、甲基纳迪克酸酐、三烷基四氢邻苯二甲酸酐、或者具有二环戊二烯骨架的酸酐或所述酸酐的改性物、羟基末端聚苯醚寡聚物、或活性酯化合物中选择。通过在本发明的含有末端改性可溶性多官能乙烯基芳香族共聚合物(xa)的硬化性组合物中并用熔融二氧化硅、结晶二氧化硅、氧化铝、氮化硅、氮化铝等无机质填充材,十溴二苯基乙烷、溴化聚苯乙烯等阻燃性赋予剂,可特别有用地用作要求介电特性或阻燃性或者耐热性的电气零件材料或电子零件材料,特别是半导体密封材料或电路基板用清漆。所述电路基板材料用清漆可通过使本发明的硬化性组合物溶解于甲苯、二甲苯、四氢呋喃、二氧杂环戊烷等有机溶剂来制造。再者,所述电路基板材料具体可列举:印刷配线基板、印刷电路板、柔性印刷配线板、增层配线板等。使本发明的含有末端改性可溶性多官能乙烯基芳香族共聚合物(xa)的硬化性组合物硬化所获得的硬化物可用作成型物、层叠物、浇铸物、粘接剂、涂膜、膜。例如,半导体密封材料的硬化物为浇铸物或成型物,作为获得所述用途的硬化物的方法,可通过利用浇铸、或转注成形机、射出成形机等来使所述化合物成形,进而在80℃~230℃下加热0.5小时~10小时来获得硬化物。另外,电路基板用清漆的硬化物为层叠物,作为获得所述硬化物的方法,可使电路基板用清漆含浸于玻璃纤维、碳纤维、聚酯纤维、聚酰胺纤维、氧化铝纤维、纸等基材中并进行加热干燥而获得预浸料,然后单独将所述预浸料彼此层叠、或与铜箔等金属箔层叠并进行热压成形而获得所述硬化物。另外,通过调配钛酸钡等无机的高介电体粉末、或铁氧体等无机磁性体,作为电子零件用材料、特别是高频电子零件材料有用。另外,与后述的硬化复合材料相同,本发明的硬化性组合物可与金属箔(包含金属板的含义。以下相同)贴合来使用。继而,对本发明的硬化性组合物的硬化性复合材料与其硬化体进行说明。为了提高机械强度、增大尺寸稳定性,而向由本发明的硬化性组合物形成的硬化性复合材料中添加基材。作为此种基材,可使用公知的基材,例如可分别单独使用纱束布、布、切股毡、表面毡等各种玻璃布,石棉布、金属纤维布及其他合成或天然的无机纤维布,自全芳香族聚酰胺纤维、全芳香族聚酯纤维、聚苯并噁唑纤维等液晶纤维所获得的织布或不织布,自聚乙烯醇纤维、聚酯纤维、丙烯酸纤维等合成纤维所获得的织布或不织布,棉布、麻布、毛毡等天然纤维布,碳纤维布、牛皮纸、棉纸、纸-玻璃混纤纸等天然纤维素系布等布类、纸类等,或者并用两种以上。在硬化性复合材料中,基材所占的比例以5wt%~90wt%,优选10wt%~80wt%,更优选20wt%~70wt%为宜。若基材少于5wt%,则复合材料的硬化后的尺寸稳定性或强度并不充分,另外,若基材多于90wt%,则复合材料的介电特性差而欠佳。在本发明的硬化性复合材料中,为了改善树脂与基材的界面的粘接性,视需要可使用偶合剂。作为偶合剂,可使用硅烷偶合剂、钛酸酯偶合剂、铝系偶合剂、铝锆偶合剂等一般的偶合剂。作为制造本发明的硬化性复合材料的方法,例如可列举如下的方法:使本发明的硬化性组合物与视需要的其他成分均匀地溶解或分散于所述芳香族系、酮系等的溶媒或其混合溶媒中,含浸于基材中后,进行干燥。含浸利用浸渍(dipping)、涂布等来进行。含浸视需要也能够重复多次,另外,此时也能够使用组成或浓度不同的多种溶液来重复含浸,并最终调整成所希望的树脂组成及树脂量。通过利用加热等方法使本发明的硬化性复合材料硬化而获得硬化复合材料。其制造方法并无特别限定,例如可使多片硬化性复合材料叠加,与在加热加压下使各层间粘接的同时进行热硬化,而获得所期望的厚度的硬化复合材料。另外,也能够将粘接硬化过一次的硬化复合材料与硬化性复合材料组合来获得新的层构成的硬化复合材料。层叠成形与硬化通常利用热压等来同时进行,但也可分别单独进行两者。即,可利用热处理或其他方法对事先进行层叠成形所获得的未硬化或半硬化的复合材料进行处理,由此使其硬化。成形及硬化可在温度:80℃~300℃、压力:0.1kg/cm2~1000kg/cm2、时间:1分钟~10小时的范围,更优选温度:150℃~250℃、压力1kg/cm2~500kg/cm2、时间:1分钟~5小时的范围内进行。本发明的层叠体是指包含本发明的硬化复合材料的层与金属箔的层者。作为此处所使用的金属箔,例如可列举铜箔、铝箔等。其厚度并无特别限定,但为3μm~200μm,更优选3μm~105μm的范围。作为制造本发明的层叠体的方法,例如可列举如下的方法:以对应于目的的层构成将以上所说明的自本发明的硬化性组合物与基材所获得的硬化性复合材料与金属箔层叠,并与在加热加压下使各层间粘接的同时进行热硬化。在本发明的硬化性组合物的层叠体中,以任意的层构成将硬化复合材料与金属箔层叠。金属箔可用作表层或中间层。除所述以外,也能够将层叠与硬化重复多次来进行多层化。与金属箔的粘接也可使用粘接剂。作为粘接剂,可列举环氧系、丙烯酸系、酚系、氰基丙烯酸酯系等,但并不特别限定于这些粘接剂。所述层叠成形与硬化可在与本发明的硬化复合材料的制造相同的条件下进行。本发明的膜是指将本发明的硬化性组合物成形为膜状而成者。其厚度并无特别限定,但为3μm~200μm,更优选5μm~100μm的范围。作为制造本发明的膜的方法,并无特别限定,例如可列举如下的方法等:使硬化性组合物与视需要的其他成分均匀地溶解或分散于芳香族系、酮系等的溶媒或其混合溶媒中,涂布于聚对苯二甲酸乙二酯(polyethyleneterephthalate,pet)膜等树脂膜上后进行干燥。涂布视需要也能够重复多次,另外,此时也能够使用组成或浓度不同的多种溶液来重复涂布,并最终调整成所希望的树脂组成及树脂量。本发明的带有树脂的金属箔是指包含本发明的硬化性组合物与金属箔者。作为此处所使用的金属箔,例如可列举铜箔、铝箔等。其厚度并无特别限定,但为3μm~200μm,更优选5μm~105μm的范围。作为制造本发明的带有树脂的金属箔的方法,并无特别限定,例如可列举如下的方法:使硬化性组合物与视需要的其他成分均匀地溶解或分散于芳香族系、酮系等的溶媒或其混合溶媒中,涂布于金属箔上后进行干燥。涂布视需要也能够重复多次,另外,此时也能够使用组成或浓度不同的多种溶液来重复涂布,并最终调整成所希望的树脂组成及树脂量。本发明的末端改性可溶性多官能乙烯基芳香族共聚合物可加工成成形材、片或膜,在电气产业、太空·飞机产业等领域中,可用于能够满足低介电常数、低吸水率、高耐热性等特性的低介电材料、绝缘材料、耐热材料、结构材料等。尤其,可用作单面、双面、多层印刷基板,柔性印刷基板,增层基板等。进而,能够应用于半导体关连材料或光学用材料,进而能够应用于涂料、感光性材料、粘接剂、污水处理剂、重金属捕获剂、离子交换树脂、抗静电剂、抗氧化剂、防雾剂、防锈剂、防染剂、杀菌剂、防虫剂、医用材料、凝聚剂、表面活性剂、润滑剂、固体燃料用粘合剂、导电处理剂等。另外,本发明的硬化性组合物提供在严酷的热历程后也具有高度的介电特性(低介电常数·低介电损耗正切)、且即便在严酷的环境下也具有高密接可靠性的硬化物,并且树脂流动性优异、低线膨胀、配线埋入平坦性优异。因此,在电气·电子产业、太空·飞机产业等领域中,可提供对应于近年来强烈要求的小型·薄型化且无翘曲等成形不良现象的硬化成形品作为介电材料、绝缘材料、耐热材料、结构材料等。进而,因配线埋入平坦性及与异种材料的密接性优异,故可实现可靠性优异的树脂组合物、硬化物或含有其的材料。以下,对本发明的硬化性树脂组合物说明第二形态。第二形态的硬化性树脂组合物尤其作为光导波管用材料有用。本发明的第二形态的硬化性树脂组合物含有(a)成分、(b)成分及(c)成分作为必需成分。此处,(a)成分为末端改性可溶性多官能乙烯基芳香族共聚合物,(b)成分为分子中具有1个以上的不饱和基的一种以上的乙烯基化合物,(c)成分为自由基聚合引发剂。用作(a)成分的末端改性可溶性多官能乙烯基芳香族共聚合物如已详细记载那样。继而,在第二形态的硬化性树脂组合物中,对(b)成分进行说明。(b)成分为分子中具有1个以上的不饱和基的一种以上的乙烯基化合物。而且,(b)成分不会与(a)成分相同。即,(a)成分不会作为(b)成分来处理。此处,乙烯基化合物只要是具有烯烃性的双键(不饱和基)的聚合性的化合物即可,不饱和基的位置并无限制,不饱和基的数量可为1个,也可为多个。以下,将不饱和基也称为乙烯基。(b)成分的乙烯基能够与(a)成分所具有的乙烯基进行共聚合。作为优选的(b)成分,有分子中具有1个以上的(甲基)丙烯酰基的一种以上的(甲基)丙烯酸酯。进而,有具有羟基和/或羧基的一种以上的(甲基)丙烯酸酯。用作(b)成分的(甲基)丙烯酸酯只要是分子中具有1个以上的(甲基)丙烯酰基者,则并无特别限制,可使用任意者。作为分子中具有1个(甲基)丙烯酰基的(甲基)丙烯酸酯化合物,例如可列举:(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸异丁酯、(甲基)丙烯酸仲丁酯、(甲基)丙烯酸叔丁酯、(甲基)丙烯酸丁氧基乙酯、(甲基)丙烯酸戊酯、(甲基)丙烯酸己酯、(甲基)丙烯酸2-乙基己酯、(甲基)丙烯酸庚酯、(甲基)丙烯酸辛酯、(甲基)丙烯酸壬酯、(甲基)丙烯酸癸酯、(甲基)丙烯酸十一酯、(甲基)丙烯酸月桂酯、(甲基)丙烯酸十三酯、(甲基)丙烯酸十四酯、(甲基)丙烯酸十五酯、(甲基)丙烯酸十六酯、(甲基)丙烯酸硬脂基酯、(甲基)丙烯酸二十二酯、甲氧基聚乙二醇(甲基)丙烯酸酯、乙氧基聚乙二醇(甲基)丙烯酸酯、甲氧基聚丙二醇(甲基)丙烯酸酯、乙氧基聚丙二醇(甲基)丙烯酸酯等脂肪族(甲基)丙烯酸酯;(甲基)丙烯酸环戊酯、(甲基)丙烯酸环己酯、(甲基)丙烯酸环戊酯、(甲基)丙烯酸二环戊酯、(甲基)丙烯酸二环戊烯酯、(甲基)丙烯酸异冰片酯等脂环式(甲基)丙烯酸酯;(甲基)丙烯酸苯酯、(甲基)丙烯酸苄酯、(甲基)丙烯酸邻联苯酯、(甲基)丙烯酸1-萘酯、(甲基)丙烯酸2-萘酯、(甲基)丙烯酸苯氧基乙酯、(甲基)丙烯酸对枯基苯氧基乙酯、(甲基)丙烯酸邻苯基苯氧基乙酯、(甲基)丙烯酸1-萘氧基乙酯、(甲基)丙烯酸2-萘氧基乙酯、苯氧基聚乙二醇(甲基)丙烯酸酯、壬基苯氧基聚乙二醇(甲基)丙烯酸酯、苯氧基聚丙二醇(甲基)丙烯酸酯等芳香族(甲基)丙烯酸酯;(甲基)丙烯酸2-四氢糠酯、n-(甲基)丙烯酰氧基乙基六氢邻苯二甲酰亚胺、2-(甲基)丙烯酰氧基乙基-n-咔唑等杂环式(甲基)丙烯酸酯等。另外,作为分子中具有2个以上的(甲基)丙烯酰基的多官能(甲基)丙烯酸酯,例如可列举:1,4-丁二醇二(甲基)丙烯酸酯、1,6-己二醇二(甲基)丙烯酸酯、1,9-壬二醇二(甲基)丙烯酸酯、三环癸烷二甲醇(甲基)丙烯酸酯、双酚a聚乙氧基二(甲基)丙烯酸酯、双酚a聚丙氧基二(甲基)丙烯酸酯、双酚f聚乙氧基二(甲基)丙烯酸酯、乙二醇二(甲基)丙烯酸酯、(甲基)丙烯酸三羟甲基丙烷三氧基乙酯、三(2-羟基乙基)异氰脲酸酯三(甲基)丙烯酸酯、季戊四醇三(甲基)丙烯酸酯、季戊四醇四(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯、聚乙二醇二(甲基)丙烯酸酯、三(丙烯酰氧基乙基)异氰脲酸酯、季戊四醇四(甲基)丙烯酸酯、二季戊四醇六(甲基)丙烯酸酯、二季戊四醇五(甲基)丙烯酸酯、三季戊四醇六(甲基)丙烯酸酯、三季戊四醇五(甲基)丙烯酸酯、羟基三甲基乙酸新戊二醇二(甲基)丙烯酸酯、羟基三甲基乙酸新戊二醇的ε-己内酯加合物的二(甲基)丙烯酸酯(例如日本化药(股份)制造的卡亚拉得(kayarad)hx-220、卡亚拉得(kayarad)hx-620等)、三羟甲基丙烷三(甲基)丙烯酸酯、三羟甲基丙烷聚乙氧基三(甲基)丙烯酸酯、二-三羟甲基丙烷四(甲基)丙烯酸酯等单体类。这些之中,就透明性及耐热性的观点而言,优选(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丙酯、(甲基)丙烯酸异丙酯、(甲基)丙烯酸丁酯、(甲基)丙烯酸2-乙基己酯等脂肪族(甲基)丙烯酸酯;脂环式(甲基)丙烯酸酯;芳香族(甲基)丙烯酸酯;杂环式(甲基)丙烯酸酯。这些化合物可单独使用、或将两种以上组合使用。当将本发明的硬化性树脂组合物设为光导波管形成用的硬化性树脂组合物时,优选包含具有由所述通式(16)或通式(17)所表示的结构单元的乙烯基化合物、或自所述乙烯基化合物所产生的硬化型乙烯基系聚合物作为(b)成分。在通式(16)或通式(17)中,r5、r8表示氢原子或甲基,r7表示氢原子或碳数1~20的一价的有机基,r6、r9表示氢原子或碳数1~20的一价的有机基。x1、x2表示可含有选自由单键、或酯键、醚键、硫酯键、硫醚键及酰胺键所组成的群组中的一种以上的键的碳数1~20的二价的有机基。具有由所述通式(16)或通式(17)所表示的结构单元的乙烯基化合物或自所述乙烯基化合物所产生的硬化型乙烯基系聚合物优选(甲基)丙烯酸酯或(甲基)丙烯酸酯系聚合物。此处,硬化型乙烯基系聚合物只要是使所述乙烯基化合物进行均聚或共聚合而获得,并具有至少一个乙烯基者即可。在通式(16)或通式(17)中,作为x1、x2为碳数1~20的二价的有机基时的二价的有机基,并无特别限制,例如可列举包含亚烷基、亚环烷基、亚苯基、亚联苯基、聚醚基、聚硅氧烷基、羰基、酯基、酰胺基、氨基甲酸酯基等的二价的有机基,这些可由卤素原子、烷基、环烷基、芳基、芳烷基、羰基、甲酰基、酯基、酰胺基、烷氧基、芳氧基、烷硫基、芳硫基、硅烷基、硅烷氧基等进一步取代。这些之中,就透明性及耐热性的观点而言,优选亚烷基、亚环烷基、亚苯基、及亚联苯基。当r7、r6、r9为碳数1~20的一价的有机基时,作为有机基,并无特别限制,例如可列举烷基、环烷基、芳基、芳烷基、酰基(-co-r)、酯基(-co-o-r或-o-co-r)、酰胺基(-co-nr2或-nr-co-r)等一价的有机基,这些可由羟基、卤素原子、烷基、环烷基、芳基、芳烷基、羰基、甲酰基、酯基、酰胺基、烷氧基、芳氧基、烷硫基、芳硫基、氨基、硅烷基、硅烷氧基等进一步取代。此处,r为烃基。这些之中,就透明性、及耐热性的观点而言,优选烷基、环烷基、芳基、及芳烷基。当(b)成分为分子中具有1个以上的(甲基)丙烯酰基的一种以上的(甲基)丙烯酸酯时,作为(甲基)丙烯酸酯,并无特别限制,例如可列举:(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸2-羟基丙酯、(甲基)丙烯酸3-氯-2-羟基丙酯、(甲基)丙烯酸2-羟基丁酯等脂肪族(甲基)丙烯酸酯,或(甲基)丙烯酸2-羟基-3-苯氧基丙酯、(甲基)丙烯酸2-羟基-3-(邻苯基苯氧基)丙酯、(甲基)丙烯酸2-羟基-3-(1-萘氧基)丙酯、(甲基)丙烯酸2-羟基-3-(2-萘氧基)丙酯等芳香族(甲基)丙烯酸酯。这些之中,就透明性及耐热性的观点而言,优选(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸2-羟基丙酯、(甲基)丙烯酸2-羟基丁酯等脂肪族(甲基)丙烯酸酯,或(甲基)丙烯酸2-羟基-3-苯氧基丙酯、(甲基)丙烯酸2-羟基-3-(邻苯基苯氧基)丙酯等芳香族(甲基)丙烯酸酯。这些化合物可单独使用、或将两种以上组合使用。当(b)成分为具有由通式(17)所表示的结构单元的乙烯基化合物或具有羧基的乙烯基化合物时,作为这些乙烯基化合物,并无特别限制,例如可列举:(甲基)丙烯酸、巴豆酸、桂皮酸、顺丁烯二酸、反丁烯二酸、柠康酸、中康酸、衣康酸、单(2-(甲基)丙烯酰氧基乙基)丁二酸酯、单(2-(甲基)丙烯酰氧基乙基)邻苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)间苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)对苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)四氢邻苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢邻苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢间苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢对苯二甲酸酯、ω-羧基-聚己内酯单(甲基)丙烯酸酯、3-乙烯基苯甲酸、4-乙烯基苯甲酸等。这些之中,就透明性、耐热性、及对于碱性显影液的溶解性的观点而言,优选(甲基)丙烯酸、巴豆酸、顺丁烯二酸、反丁烯二酸、单(2-(甲基)丙烯酰氧基乙基)丁二酸酯、单(2-(甲基)丙烯酰氧基乙基)四氢邻苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢邻苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢间苯二甲酸酯、单(2-(甲基)丙烯酰氧基乙基)六氢对苯二甲酸酯。另外,也可适宜地使用利用甲醇、乙醇、丙醇等适当的醇对顺丁烯二酸酐、柠康酸酐等具有2个以上的羧基的乙烯基化合物或其酸酐的一部分的羧基进行部分酯化而成的乙烯基化合物。另外,也可适宜地使用使具有2个以上的羧基的乙烯基化合物的酸酐与分子中具有1个以上的(甲基)丙烯酰基的(甲基)丙烯酸酯化合物进行共聚合后,利用甲醇、乙醇、丙醇等适当的醇进行部分酯化而成的乙烯基化合物。这些化合物可单独使用、或将两种以上组合使用。在光导波管形成用的硬化性树脂组合物中,可优选地使用具有氨基甲酸酯键与1个以上的(甲基)丙烯酰基的硬化型(甲基)丙烯酸酯(b1)作为(b)成分的乙烯基化合物。作为所述硬化型(甲基)丙烯酸酯(b1),并无特别限制,例如可列举下述(1)~下述(4)的(甲基)丙烯酸氨基甲酸酯等。(1)使二官能醇化合物、二官能异氰酸酯化合物、及具有羟基的(甲基)丙烯酸酯进行反应所获得的(甲基)丙烯酸氨基甲酸酯。(2)使二官能醇化合物、二官能异氰酸酯化合物、及具有异氰酸酯基的(甲基)丙烯酸酯进行反应所获得的(甲基)丙烯酸氨基甲酸酯。(3)使多官能异氰酸酯化合物及具有羟基的(甲基)丙烯酸酯进行反应所获得的(甲基)丙烯酸氨基甲酸酯。(4)使多官能醇化合物及具有异氰酸酯基的(甲基)丙烯酸酯进行反应所获得的(甲基)丙烯酸氨基甲酸酯。这些之中,就透明性及耐热性的观点而言,优选的是其分子中具有选自由脂环结构、芳香环结构、及杂环结构所组成的群组中的至少一种的(甲基)丙烯酸氨基甲酸酯。作为所述二官能醇化合物,即二醇化合物,并无特别限制,例如可列举:聚醚二醇化合物、聚酯二醇化合物、聚碳酸酯二醇化合物、聚己内酯二醇化合物、其他二醇化合物等。作为所述聚醚二醇化合物,并无特别限制,例如可列举:通过使选自环氧乙烷、环氧丙烷、氧化异丁烯、丁基缩水甘油醚、丁烯-1-氧化物、3,3-双氯甲基氧杂环丁烷、四氢呋喃、2-甲基四氢呋喃、3-甲基四氢呋喃、环氧环己烷、氧化苯乙烯、苯基缩水甘油醚、苯甲酸缩水甘油酯等环状醚化合物中的至少一种进行开环(共)聚合所获得的聚醚二醇化合物;通过使环己烷二甲醇、三环癸烷二甲醇、氢化双酚a、氢化双酚f等脂环式二醇化合物与选自所述环状醚化合物中的至少一种进行开环加成所获得的聚醚二醇化合物,或通过使对苯二酚、间苯二酚、儿茶酚、双酚a、双酚f、双酚af、联苯酚、芴双酚等二官能酚化合物与选自所述环状醚化合物中的至少一种进行开环加成所获得的聚醚二醇化合物等。作为所述聚酯二醇化合物,并无特别限制,例如可列举:使丙二酸、丁二酸、戊二酸、己二酸、癸二酸、四氢邻苯二甲酸、六氢邻苯二甲酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、顺丁烯二酸、反丁烯二酸、衣康酸等二官能羧酸化合物与乙二醇、二乙二醇、聚乙二醇、丙二醇、二丙二醇、聚丙二醇、丁二醇、二丁二醇、聚丁二醇、戊二醇、新戊二醇、3-甲基-1,5-戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇、环己烷二甲醇、三环癸烷二甲醇等二醇化合物进行共聚合所获得的聚酯多元醇化合物等。作为所述聚碳酸酯二醇化合物,并无特别限制,例如可列举:使碳酰氯、三碳酰氯、碳酸二烷基酯、碳酸二芳基酯等与乙二醇、二乙二醇、聚乙二醇、丙二醇、二丙二醇、聚丙二醇、丁二醇、二丁二醇、聚丁二醇、戊二醇、新戊二醇、3-甲基-1,5-戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇、环己烷二甲醇、三环癸烷二甲醇、氢化双酚a、氢化双酚f等二醇化合物进行共聚合所获得的聚碳酸酯二醇化合物等。作为所述聚己内酯二醇化合物,并无特别限制,例如可列举:使ε-己内酯与乙二醇、二乙二醇、聚乙二醇、丙二醇、二丙二醇、聚丙二醇、丁二醇、二丁二醇、聚丁二醇、戊二醇、新戊二醇、3-甲基-1,5-戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇、环己烷二甲醇、三环癸烷二甲醇等二醇化合物进行共聚合所获得的聚己内酯二醇化合物等。作为其他二醇化合物,例如可列举:乙二醇、二乙二醇、丙二醇、二丙二醇、丁二醇、二丁二醇、戊二醇、新戊二醇、3-甲基-1,5-戊二醇、己二醇、庚二醇、辛二醇、壬二醇、癸二醇等脂肪族二醇化合物;环己烷二甲醇、三环癸烷二甲醇、氢化双酚a、氢化双酚f等脂环式二醇化合物;聚丁二烯改性二醇化合物、氢化聚丁二烯改性二醇化合物、硅酮改性二醇化合物等改性二醇化合物等。这些二醇化合物可单独使用、或将两种以上组合使用。作为所述二官能异氰酸酯化合物,并无特别限制,例如可列举:四亚甲基二异氰酸酯、六亚甲基二异氰酸酯、2,2,4-三甲基六亚甲基二异氰酸酯、十亚甲基二异氰酸酯、十二亚甲基二异氰酸酯等脂肪族二官能异氰酸酯化合物;1,3-双(异氰酸基甲基)环己烷、异佛尔酮二异氰酸酯、2,5-双(异氰酸基甲基)降冰片烯、双(4-异氰酸基环己基)甲烷、1,2-双(4-异氰酸基环己基)乙烷、2,2-双(4-异氰酸基环己基)丙烷、2,2-双(4-异氰酸基环己基)六氟丙烷、二环庚烷三异氰酸酯等脂环式二官能异氰酸酯化合物;2,4'-二苯基甲烷二异氰酸酯、4,4'-二苯基甲烷二异氰酸酯、2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯、萘-1,5-二异氰酸酯、邻亚二甲苯基二异氰酸酯、间亚二甲苯基二异氰酸酯等芳香族二官能异氰酸酯化合物等。这些化合物可单独使用、或将两种以上组合使用。作为所述具有羟基的(甲基)丙烯酸酯,并无特别限制,例如可列举:(甲基)丙烯酸2-羟基乙酯、(甲基)丙烯酸2-羟基丙酯、(甲基)丙烯酸3-氯-2-羟基丙酯、(甲基)丙烯酸2-羟基丁酯、(甲基)丙烯酸2-羟基-3-苯氧基丙酯、(甲基)丙烯酸2-羟基-3-(邻苯基苯氧基)丙酯、(甲基)丙烯酸2-羟基-3-(1-萘氧基)丙酯、(甲基)丙烯酸2-羟基-3-(2-萘氧基)丙酯等单官能(甲基)丙烯酸酯,这些的乙氧基化体,这些的丙氧基化体,这些的乙氧基化丙氧基化体,及这些的己内酯改性体;双(2-(甲基)丙烯酰氧基乙基)(2-羟基乙基)异氰脲酸酯等二官能(甲基)丙烯酸酯、这些的乙氧基化体、这些的丙氧基化体、这些的乙氧基化丙氧基化体、及这些的己内酯改性体;环己烷二甲醇型环氧基二(甲基)丙烯酸酯、三环癸烷二甲醇型环氧基二(甲基)丙烯酸酯、氢化双酚a型环氧基二(甲基)丙烯酸酯、氢化双酚f型环氧基二(甲基)丙烯酸酯、对苯二酚型环氧基二(甲基)丙烯酸酯、间苯二酚型环氧基二(甲基)丙烯酸酯、儿茶酚型环氧基二(甲基)丙烯酸酯、双酚a型环氧基二(甲基)丙烯酸酯、双酚f型环氧基二(甲基)丙烯酸酯、双酚af型环氧基二(甲基)丙烯酸酯、联苯酚型环氧基二(甲基)丙烯酸酯、芴双酚型环氧基二(甲基)丙烯酸酯、异氰脲酸单烯丙基型环氧基二(甲基)丙烯酸酯等二官能环氧(甲基)丙烯酸酯;季戊四醇三(甲基)丙烯酸酯、二季戊四醇四(甲基)丙烯酸酯、二季戊四醇五(甲基)丙烯酸酯等三官能以上的(甲基)丙烯酸酯,这些的乙氧基化体,这些的丙氧基化体,这些的乙氧基化丙氧基化体,及这些的己内酯改性体;苯酚酚醛清漆型环氧(甲基)丙烯酸酯、甲酚酚醛清漆型环氧基聚(甲基)丙烯酸酯、异氰脲酸型环氧基三(甲基)丙烯酸酯等三官能以上的环氧(甲基)丙烯酸酯等。这些化合物可单独使用、或将两种以上组合使用。此处,所谓(甲基)丙烯酸酯的乙氧基化体、丙氧基化体、乙氧基化丙氧基化体,表示代替成为原料的醇化合物或酚化合物(例如,在单官能(甲基)丙烯酸酯;ch2=ch(r7)-coo-r8(r7为氢原子或甲基,r8为一价的有机基)的情况下,由ho-r8所表示者),将在所述醇化合物或酚化合物中分别加成1个以上的环氧乙烷而成的结构的醇化合物、加成1个以上的环氧丙烷而成的结构的醇化合物、或加成1个以上的环氧乙烷及环氧丙烷而成的结构的醇化合物用于原料所获得的(甲基)丙烯酸酯(例如,在乙氧基化体的情况下由ch2=ch(r7)-coo-(ch2ch2o)q-r8(q为1以上的整数,r7、r8与所述相同)表示)。另外,所谓己内酯改性体,表示将利用ε-己内酯对成为(甲基)丙烯酸酯的原料的醇化合物进行改性而成的醇化合物用于原料所获得的(甲基)丙烯酸酯(例如,在单官能(甲基)丙烯酸酯的己内酯改性体的情况下,由ch2=ch(r7)-coo-((ch2)5coo)q-r8(q、r7、r8与所述相同)表示)。作为所述具有异氰酸酯基的(甲基)丙烯酸酯,并无特别限制,例如可列举:n-(甲基)丙烯酰基异氰酸酯、(甲基)丙烯酰氧基甲基异氰酸酯、2-(甲基)丙烯酰氧基乙基异氰酸酯、2-(甲基)丙烯酰氧基乙氧基乙基异氰酸酯、1,1-双((甲基)丙烯酰氧基甲基)乙基异氰酸酯等。这些化合物可单独使用、或将两种以上组合使用。作为所述多官能异氰酸酯化合物,并无特别限制,例如可列举:所述二官能异氰酸酯化合物;所述二官能异氰酸酯化合物的脲二酮型二聚体、异氰脲酸酯型三聚体、缩二脲型三聚体等多聚体等。再者,构成多聚体的两种或三种二官能异氰酸酯化合物可相同,也可不同。这些化合物可单独使用、或将两种以上组合使用。作为所述多官能醇化合物,并无特别限制,例如可列举:所述二官能醇化合物;三羟甲基丙烷、季戊四醇、二-三羟甲基丙烷、二季戊四醇、三(2-羟基乙基)异氰脲酸酯等三官能以上的醇化合物,通过使这些与选自所述环状醚化合物中的至少一种进行开环加成所获得的加合物,这些的己内酯改性体;通过使苯酚酚醛清漆、甲酚酚醛清漆等三官能以上的酚化合物与选自所述环状醚化合物中的至少一种进行开环加成所获得的醇化合物,这些的己内酯改性体。这些化合物可单独使用、或将两种以上组合使用。就耐热性及对于碱性显影液的溶解性的观点而言,所述具有氨基甲酸酯键的硬化型(甲基)丙烯酸酯视需要可进而含有羧基。作为所述具有羧基与氨基甲酸酯键的(甲基)丙烯酸酯,并无特别限制,例如可列举:在合成所述(甲基)丙烯酸氨基甲酸酯时,将含有羧基的二醇化合物与所述二醇化合物并用、或代替所述二醇化合物而使用含有羧基的二醇化合物所获得的(甲基)丙烯酸氨基甲酸酯等。作为含有羧基的二醇化合物,并无特别限制,例如可列举:2,2-二羟甲基丁酸、2,2-二羟甲基丙酸、2,2-二羟甲基丁酸、2,2-二羟甲基戊酸等。这些化合物可单独使用、或将两种以上组合使用。所述具有羧基与氨基甲酸酯键的硬化型(甲基)丙烯酸酯能够以可利用后述的碱性显影液进行显影的方式规定酸值。酸值优选5mgkoh/g~200mgkoh/g,更优选10mgkoh/g~170mgkoh/g,特优选15mgkoh/g~150mgkoh/g。若为5mgkoh/g以上,则对于碱性显影液的溶解性良好,若为200mgkoh/g以下,则耐显影液性良好。在光导波管形成用的硬化性树脂组合物中,优选的是进而使用多官能封闭型异氰酸酯化合物作为(f)成分。(f)成分与具有羟基和/或羧基的成分进行反应而产生交联结构。在此情况下,(b)成分优选的是具有羟基或羧基的乙烯基化合物,当不具有(b)成分时,理想的是调配产生所述反应的具有羟基或羧基的化合物(e)。作为(f)成分的多官能封闭型异氰酸酯化合物为利用多官能异氰酸酯化合物与封闭剂的反应所生成的化合物。另外,其为利用源自封闭剂的基而暂时钝化的化合物,若加热至规定温度,则源自封闭剂的基解离,并生成异氰酸酯基。若使用多官能封闭型异氰酸酯化合物,则使因加热而自多官能封闭型异氰酸酯化合物中生成的异氰酸酯基与(a)成分的羟基和/或羧基进行反应,由此可形成新的交联结构,并提升耐热性及环境可靠性。作为可与封闭剂进行反应的多官能异氰酸酯化合物,并无特别限制,例如可列举:2,4'-二苯基甲烷二异氰酸酯、4,4'-二苯基甲烷二异氰酸酯、2,4-甲苯二异氰酸酯、2,6-甲苯二异氰酸酯、萘-1,5-二异氰酸酯、邻亚二甲苯基二异氰酸酯、间亚二甲苯基二异氰酸酯等芳香族多官能异氰酸酯化合物;1,3-双(异氰酸基甲基)环己烷、异佛尔酮二异氰酸酯、2,5-双(异氰酸基甲基)降冰片烯、双(4-异氰酸基环己基)甲烷、1,2-双(4-异氰酸基环己基)乙烷、2,2-双(4-异氰酸基环己基)丙烷、2,2-双(4-异氰酸基环己基)六氟丙烷、二环庚烷三异氰酸酯等脂环式多官能异氰酸酯化合物;四亚甲基二异氰酸酯、六亚甲基二异氰酸酯、2,2,4-三甲基六亚甲基二异氰酸酯、十亚甲基二异氰酸酯、十二亚甲基二异氰酸酯等脂肪族多官能异氰酸酯化合物等。作为多官能异氰酸酯化合物,就透明性及耐热性的观点而言,优选其分子中含有选自脂环结构及脂肪族结构中的至少一种的化合物,其中,优选所述脂环式多官能异氰酸酯化合物;所述脂肪族多官能异氰酸酯化合物。再者,所述多官能异氰酸酯化合物可为脲二酮型二聚体、异氰脲酸酯型三聚体、或缩二脲型三聚体等多聚体,构成这些多聚体的两种或三种多官能异氰酸酯化合物可相同,也可不同。另外,在不同的情况下,例如可如脂环式多官能异氰酸酯化合物与脂肪族多官能异氰酸酯化合物的组合那样为不同种类的多官能异氰酸酯化合物的组合。以上的多官能异氰酸酯化合物可单独使用、或将两种以上组合使用。作为封闭剂,优选的是具有活性氢者,例如可列举:丙二酸二酯、乙酰乙酸酯、丙二酸二腈、乙酰丙酮、亚甲基二砜、二苯甲酰基甲烷、二三甲基乙酰基甲烷、丙酮二羧酸二酯等活性亚甲基化合物;丙酮肟、甲基乙基酮肟、二乙基酮肟、甲基异丁基酮肟、环己酮肟等肟化合物;苯酚、烷基苯酚、烷基萘酚等酚化合物;γ-丁内酰胺、δ-戊内酰胺、ε-己内酰胺等内酰胺化合物等。这些之中,就透明性及耐热性的观点而言,优选所述活性亚甲基化合物;所述肟化合物;所述内酰胺化合物。以上的封闭剂可单独使用、或将两种以上组合使用。在本发明的硬化性树脂组合物中,用作(c)成分的自由基聚合引发剂通过加热或紫外线、可见光线等光化射线的照射,而使(a)成分及(b)成分的自由基聚合开始。作为自由基聚合引发剂,只要是通过加热或紫外线、可见光线等光化射线的照射而使自由基聚合开始者,则并无特别限制,例如可列举:热自由基聚合引发剂、光自由基聚合引发剂等。作为热自由基聚合引发剂,并无特别限制,例如可列举:甲基乙基酮过氧化物、环己酮过氧化物、甲基环己酮过氧化物等酮过氧化物;1,1-双(过氧化叔丁基)环己烷、1,1-双(过氧化叔丁基)-2-甲基环己烷、1,1-双(过氧化叔丁基)-3,3,5-三甲基环己烷、1,1-双(过氧化叔己基)环己烷、1,1-双(过氧化叔己基)-3,3,5-三甲基环己烷等过氧缩酮;对薄荷烷氢过氧化物等氢过氧化物;α,α'-双(过氧化叔丁基)二异丙基苯、二枯基过氧化物、叔丁基枯基过氧化物、二-叔丁基过氧化物等二烷基过氧化物;辛酰基过氧化物、月桂酰基过氧化物、硬脂基过氧化物、苯甲酰基过氧化物等二酰基过氧化物;过氧化二碳酸双(4-叔丁基环己基)酯、过氧化二碳酸二-2-乙氧基乙酯、过氧化二碳酸二-2-乙基己酯、过氧化碳酸二-3-甲氧基丁酯等过氧化碳酸酯;过氧化三甲基乙酸叔丁酯、过氧化三甲基乙酸叔己酯、过氧化-2-乙基己酸1,1,3,3-四甲基丁酯、2,5-二甲基-2,5-双(过氧化2-乙基己酰基)己烷、过氧化-2-乙基己酸叔己酯、过氧化-2-乙基己酸叔丁酯、过氧化异丁酸叔丁酯、过氧化异丙基单碳酸叔己酯、过氧化-3,5,5-三甲基己酸叔丁酯、过氧化月桂酸叔丁酯、过氧化异丙基单碳酸叔丁酯、过氧化-2-乙基己基单碳酸叔丁酯、过氧化苯甲酸叔丁酯、过氧化苯甲酸叔己酯、2,5-二甲基-2,5-双(过氧化苯甲酰基)己烷、过氧化乙酸叔丁酯等过氧酯;2,2'-偶氮双异丁腈、2,2'-偶氮双(2,4-二甲基戊腈)、2,2'-偶氮双(4-甲氧基-2'-二甲基戊腈)等偶氮化合物。这些之中,就硬化性、透明性、及耐热性的观点而言,优选二酰基过氧化物、过氧酯、及偶氮化合物。作为光自由基聚合引发剂,只要是通过紫外线、可见光线等光化射线的照射而使自由基聚合开始者,则并无特别限制,例如可列举:2,2-二甲氧基-1,2-二苯基乙烷-1-酮等安息香缩酮;1-羟基环己基苯基酮、2-羟基-2-甲基-1-苯基丙烷-1-酮、1-[4-(2-羟基乙氧基)苯基]-2-羟基-2-甲基-1-丙烷-1-酮、2-羟基-1-{4[4-(2-羟基-2-甲基丙酰基)苄基]苯基}-2-甲基丙烷-1-酮等α-羟基酮;苯基乙醛酸甲酯、苯基乙醛酸乙酯、氧基苯基乙酸2-(2-羟基乙氧基)乙酯、氧基苯基乙酸2-(2-侧氧基-2-苯基乙酰氧基乙氧基)乙酯等乙醛酸酯;2-苄基-2-二甲基氨基-1-(4-吗啉-4-基苯基)-丁烷-1-酮、2-二甲基氨基-2-(4-甲基苄基)-1-(4-吗啉-4-基苯基)-丁烷-1-酮、1,2-甲基-1-[4-(甲硫基)苯基]-(4-吗啉)-2-基丙烷-1-酮等α-氨基酮;1,2-辛二酮,1-[4-(苯硫基),2-(o-苯甲酰基肟)]、乙酮,1-[9-乙基-6-(2-甲基苯甲酰基)-9h-咔唑-3-基],1-(o-乙酰基肟)等肟酯;双(2,4,6-三甲基苯甲酰基)苯基氧化膦、双(2,6-二甲氧基苯甲酰基)-2,4,4-三甲基戊基氧化膦、2,4,6-三甲基苯甲酰基二苯基氧化膦等氧化膦;2-(邻氯苯基)-4,5-二苯基咪唑二聚体、2-(邻氯苯基)-4,5-二(甲氧基苯基)咪唑二聚体、2-(邻氟苯基)-4,5-二苯基咪唑二聚体、2-(邻甲氧基苯基)-4,5-二苯基咪唑二聚体、2-(对甲氧基苯基)-4,5-二苯基咪唑二聚体等2,4,5-三芳基咪唑二聚体;二苯甲酮、n,n'-四甲基-4,4'-二氨基二苯甲酮、n,n'-四乙基-4,4'-二氨基二苯甲酮、4-甲氧基-4'-二甲基氨基二苯甲酮等二苯甲酮化合物;2-乙基蒽醌、菲醌、2-叔丁基蒽醌、八甲基蒽醌、1,2-苯并蒽醌、2,3-苯并蒽醌、2-苯基蒽醌、2,3-二苯基蒽醌、1-氯蒽醌、2-甲基蒽醌、1,4-萘醌、9,10-菲醌、2-甲基-1,4-萘醌、2,3-二甲基蒽醌等醌化合物;安息香甲基醚、安息香乙基醚、安息香苯基醚等安息香醚;安息香、甲基安息香、乙基安息香等安息香化合物;苄基二甲基缩酮等苄基化合物;9-苯基吖啶、1,7-双(9,9'-吖啶基庚烷)等吖啶化合物;n-苯基甘胺酸、香豆素等。此处,2,4,5-三芳基咪唑二聚体的2个三芳基咪唑部位的芳基的取代基可相同而提供对称的化合物,也可不同而提供非对称的化合物。所述光自由基聚合引发剂中,就硬化性及透明性的观点而言,优选如上所述的α-羟基酮、乙醛酸酯、肟酯或氧化膦。以上的自由基聚合引发剂(热自由基聚合引发剂及光自由基聚合引发剂等)可单独使用、或将两种以上组合使用,进而也可与适当的敏化剂组合使用。在本发明的硬化性树脂组合物中,所述(a)成分、(b)成分、及(c)成分的调配量是(a)成分为5wt%~94.9wt%,(b)成分为5.0wt%~85wt%,及(c)成分为0.1wt%~10wt%。优选(a)成分为30wt%~94.9wt%,(b)成分为5.0wt%~60wt%,及(c)成分为0.1wt%~10wt%,更优选(a)成分为40wt%~80wt%,(b)成分为10wt%~50wt%,及(c)成分为1wt%~5wt%。在本发明的硬化性树脂组合物中,视需要可调配溶剂、其他添加剂。而且,在硬化性树脂组合物中的调配量的计算中,硬化后被去除的溶剂等的挥发成分自计算中排除。当调配添加剂时,所述(a)成分、(b)成分、及(c)成分的调配量相对于(a)成分、(b)成分、及(c)成分的合计,(a)成分为5wt%~94.9wt%,(b)成分为5.0wt%~85wt%,及(c)成分为0.1wt%~10wt%。优选(a)成分为30wt%~94.9wt%,(b)成分为5.0wt%~60wt%,及(c)成分为0.1wt%~10wt%,更优选(a)成分为40wt%~80wt%,(b)成分为10wt%~50wt%,及(c)成分为1wt%~5wt%。另外,在本发明的光导波管形成用的硬化性树脂组合物中,(a)成分、(b)成分、及(c)成分的调配量以(a)成分为10wt%~70wt%,(b)成分为15wt%~70wt%,及(c)成分为0.1wt%~10wt%为宜。进而,理想的是调配作为(f)成分的多官能封闭型异氰酸酯化合物及作为(e)成分的具有羟基或羧基的化合物,这些化合物的调配量是(f)成分为10wt%~40wt%,(e)成分为0wt%~40wt%。此处,(b)成分以所述(b1)成分为宜,进而以在(b)成分中含有10wt%~70wt%的具有羟基或羧基的乙烯基(b2)为宜。当(e)成分为具有羟基或羧基的乙烯基化合物时,作为相当于(b)成分及(e)成分两者的成分进行计算。就其他观点而言,当不含(e)成分时,相对于(a)成分~(c)成分的总量,(f)成分的调配量优选1质量%~40质量%。另外,当含有(e)成分时,相对于(a)成分~(c)成分及(e)成分的总量,(f)成分的调配量优选1质量%~40质量%。若(f)成分的调配量为所述范围,则(e)成分的羟基和/或羧基与自多官能封闭型异氰酸酯化合物所生成的异氰酸酯基进行反应而形成充分的交联结构,因此可获得耐热性良好、强韧性良好且不会变脆的硬化物。就以上的观点而言,(f)成分的调配量更优选3质量%~35质量%,特优选5质量%~30质量%。在光导波管形成用的硬化性树脂组合物的情况下,当使用所述具有氨基甲酸酯键的(甲基)丙烯酸酯(b1)作为(b)成分时,可将具有氨基甲酸酯键的(甲基)丙烯酸酯(b1)设为(b)成分的全部,但视需要也可使用其他乙烯基化合物。在本发明的树脂组合物或光导波管形成用的树脂组合物中,视需要能够以不对本发明的效果造成不良影响的比例,进而添加抗氧化剂、抗黄变剂、紫外线吸收剂、可见光吸收剂、着色剂、塑化剂、稳定剂、填充剂等所谓的添加剂。另外,这些树脂组合物也可使用适当的有机溶剂进行稀释,而用作树脂清漆。作为此处所使用的有机溶剂,只要是可溶解树脂组合物者,则并无特别限制,例如可列举:甲苯、二甲苯、1,3,5-三甲苯、枯烯、对异丙基甲苯等芳香族烃;二乙基醚、叔丁基甲基醚、环戊基甲基醚、二丁基醚等链状醚;四氢呋喃、1,4-二噁烷等环状醚;甲醇、乙醇、异丙醇、丁醇、乙二醇、丙二醇等醇;丙酮、甲基乙基酮、甲基异丁基酮、环己酮、4-羟基-4-甲基-2-戊酮等酮;乙酸甲酯、乙酸乙酯、乙酸丁酯、乳酸甲酯、乳酸乙酯、γ-丁内酯等酯;碳酸亚乙酯、碳酸亚丙酯等碳酸酯;乙二醇单甲基醚、乙二醇单乙基醚、乙二醇单丁基醚、乙二醇二甲基醚、乙二醇二乙基醚、丙二醇单甲基醚、丙二醇单乙基醚、丙二醇二甲基醚、丙二醇二乙基醚、二乙二醇单甲基醚、二乙二醇单乙基醚、二乙二醇单丁基醚、二乙二醇二甲基醚、二乙二醇二乙基醚等二官能醇烷基醚;乙二醇单甲基醚乙酸酯、乙二醇单乙基醚乙酸酯、乙二醇单丁基醚乙酸酯、丙二醇单甲基醚乙酸酯、丙二醇单乙基醚乙酸酯、二乙二醇单甲基醚乙酸酯、二乙二醇单乙基醚乙酸酯等二官能醇烷基醚乙酸酯;n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、n-甲基吡咯烷酮等酰胺等。这些有机溶剂可单独使用、或将两种以上组合使用。另外,树脂清漆中的固体成分浓度通常优选10质量%~80质量%。当将本发明的树脂组合物或光导波管形成用树脂组合物制成清漆时,优选通过搅拌来混合。搅拌方法并无特别限制,但就搅拌效率的观点而言,优选使用螺旋桨的搅拌。进行搅拌时的螺旋桨的旋转速度并无特别限制,但优选10rpm~1,000rpm。若螺旋桨的旋转速度为10rpm以上,则各成分得到充分混合,若为1,000rpm以下,则由螺旋桨的旋转所产生的气泡的卷入变少。就以上的观点而言,螺旋桨的旋转速度更优选50rpm~800rpm,特优选100rpm~500rpm。另外,搅拌时间并无特别限制,但优选1小时~24小时。若搅拌时间为1小时以上,则各成分得到充分混合,若为24小时以下,则可缩短调合时间,生产性提升。所述清漆优选使用孔径为50μm以下的过滤器进行过滤。通过使用孔径为50μm以下的过滤器,大的异物等被去除且在涂布时不会产生凹陷等,另外,光的散射得到抑制且透明性不会受损。就以上的观点而言,更优选使用孔径为30μm以下的过滤器进行过滤,特优选使用孔径为10μm以下的过滤器进行过滤。另外,在为经调合的树脂清漆、或并非清漆的树脂组合物,且含有挥发成分时,优选的是在减压下进行脱泡。脱泡方法并无特别限制,例如可列举:使用真空泵与钟罩、带有真空装置的脱泡装置的方法。减压时的压力并无特别限制,但优选的是树脂组合物中所含有的低沸点成分不会沸腾的压力。减压脱泡时间并无特别限制,但优选3分钟~60分钟。若减压脱泡时间为3分钟以上,则可去除溶解于树脂组合物内的气泡,若为60分钟以下,则树脂组合物中所含有的有机溶剂不会挥发,且可缩短脱泡时间,而可提升生产性。继而,对本发明的光导波管形成用树脂膜进行说明。所述树脂膜使用所述光导波管形成用的树脂组合物来形成。例如,可通过将光导波管形成用树脂组合物涂布于适当的支撑膜上而容易地制造。另外,当所述树脂组合物为经有机溶剂稀释的树脂清漆时,可通过将树脂清漆涂布于支撑膜上,并去除有机溶剂来制造。作为支撑膜,并无特别限制,例如可列举:聚对苯二甲酸乙二酯、聚对苯二甲酸丁二酯、聚萘二甲酸乙二酯等聚酯;聚乙烯、聚丙烯等聚烯烃;聚碳酸酯、聚酰胺、聚酰亚胺、聚酰胺酰亚胺、聚醚酰亚胺、聚醚硫化物、聚醚砜、聚醚酮、聚苯醚、聚苯硫醚、聚芳酯、聚砜、液晶聚合物等。这些之中,就柔软性及强韧性的观点而言,优选聚对苯二甲酸乙二酯、聚对苯二甲酸丁二酯、聚萘二甲酸乙二酯、聚丙烯、聚碳酸酯、聚酰胺、聚酰亚胺、聚酰胺酰亚胺、聚苯醚、聚苯硫醚、聚芳酯、聚砜。再者,就提升与树脂层的剥离性的观点而言,视需要可使用通过硅酮系化合物、含氟化合物等实施了脱模处理的膜。支撑膜的厚度可根据作为目标的柔软性而适宜变更,但就膜强度与柔软性的观点而言,优选3μm~250μm,更优选5μm~200μm,特优选7μm~150μm。将光导波管形成用的树脂组合物涂布于支撑膜上而成的膜视需要可在树脂层上贴附保护膜,而制成包含支撑膜、树脂层、及保护膜的3层结构。作为保护膜,并无特别限制,但就柔软性及强韧性的观点而言,优选聚对苯二甲酸乙二酯、聚对苯二甲酸丁二酯、聚萘二甲酸乙二酯等聚酯;聚乙烯、聚丙烯等聚烯烃等。再者,就提升与树脂层的剥离性的观点而言,视需要可使用通过硅酮系化合物、含氟化合物等实施了脱模处理的膜。保护膜的厚度优选10μm~250μm,更优选15μm~200μm,特优选20μm~150μm。本发明的光导波管形成用树脂膜的树脂层的厚度也无特别限制,但以干燥后的厚度计通常优选5μm~500μm。若为所述范围内,则树脂膜或树脂膜的硬化物的强度充分,同时干燥可充分地进行,因此树脂膜中的残留溶剂量不会增加,当对树脂膜的硬化物进行加热时不会发泡。以所述方式获得的光导波管形成用树脂膜例如可通过卷取成卷状而容易地保存。另外,也可将卷状的膜切成所期望的尺寸,而变成片状来保存。继而,对将本发明的光导波管形成用树脂膜作为芯部形成用的树脂膜时的应用例进行说明。作为所述情况下的支撑膜,只要是用于芯图案形成的曝光用光化射线透过者,则并无特别限制,例如可适宜地列举与作为所述光导波管形成用树脂膜的支撑膜的具体例所记载者相同的支撑膜。这些之中,就曝光用光化射线的透过率、柔软性、及强韧性的观点而言,优选聚酯;聚烯烃。进而,就提升曝光用光化射线的透过率及降低芯图案的侧壁粗糙度的观点而言,更优选使用高透明型的支撑膜。作为此种高透明型的支撑膜,在市售者中,例如可列举:东洋纺(股份)制造的“克斯莫闪(cosmoshine)a1517”、“克斯莫闪(cosmoshine)a4100”,东丽(股份)制造的“露米勒(lumirror)fb50”等。就强度与芯图案形成时的图案分辨率的观点而言,所述支撑膜的厚度优选5μm~50μm,更优选10μm~40μm,特优选15μm~30μm。继而,对将本发明的光导波管形成用树脂膜作为包层形成用树脂膜(上部包层形成用树脂膜、下部包层形成用树脂膜)的情况进行说明。作为所述情况下的支撑膜,只要是用于包覆形成的曝光用光化射线透过者,则并无特别限制,例如可适宜地列举与作为所述光导波管形成用树脂膜的支撑膜的具体例所记载者相同的支撑膜。这些之中,就曝光用光化射线的透过率、柔软性、及强韧性的观点而言,优选聚酯;聚烯烃。进而,就提升曝光用光化射线的透过率及降低包覆图案的侧壁粗糙度的观点而言,更优选使用高透明型的支撑膜。作为此种高透明型的支撑膜,在市售者中,例如可列举:东洋纺(股份)制造的“克斯莫闪(cosmoshine)a1517”、“克斯莫闪(cosmoshine)a4100”,东丽(股份)制造的“露米勒(lumirror)fb50”等。包层形成用树脂膜的支撑膜的厚度优选5μm~50μm,更优选10μm~40μm,特优选15μm~30μm继而,对本发明的光导波管进行说明。图1的(a)~(d)分别表示光导波管的剖面图。在图1的(a)中,光导波管1形成于基材5上,包括包含高折射率的芯部形成用树脂组合物的芯部2、以及包含低折射率的包层形成用树脂组合物的下部包层4及上部包层3。图1的(b)~(d)分别表示其他形态。(b)表示在上部包层3的外侧配置有基材5作为保护膜的形态。(c)表示在下部包层4及上部包层3两者的外侧配置有基材5作为保护膜的形态。如(d),表示未配置作为保护膜5的基材的形态。本发明的光导波管形成用的树脂组合物及光导波管形成用树脂膜以用于光导波管1的下部包层4、芯部2、及上部包层3中的至少一者为宜,优选的是以将调整了折射率的这些用于所有层为宜。通过使用光导波管形成用树脂膜,可进一步提升各层的平坦性、包覆与芯的层间密接性、及光导波管芯图案形成时的分辨率(细线或窄间隙对应性),能够形成平坦性优异、线宽或间隙小的微细图案。在光导波管1中,作为基材5的材质,并无特别限制,例如可列举:玻璃环氧树脂基板、陶瓷基板、玻璃基板、硅基板、塑料基板、金属基板、带有树脂层的基板、带有金属层的基板、塑料膜、带有树脂层的塑料膜、带有金属层的塑料膜等。光导波管1通过将作为基材5的具有柔软性及强韧性的基材,例如所述光导波管形成用树脂膜的支撑膜用作基材,而可制成柔性光导波管,此时也可使基材5作为光导波管1的保护膜发挥功能。通过配置保护膜,能够对光导波管1赋予保护膜的柔软性及强韧性。进而,光导波管1不会受到污染或损伤,因此处理容易性提升。就以上的观点而言,有时如(b)~(c)那样的形态适合代替图1的(a)的形态。再者,若光导波管的柔软性或强韧性充分,则如(d)的那样,能够省略保护膜。下部包层4的厚度并无特别限制,但优选2μm~200μm。若为2μm以上,则容易将传播光封入至芯内部,若为200μm以下,则光导波管1整体的厚度不会过大。再者,所谓下部包层4的厚度,是指自芯部2与下部包层4的边界至下部包层4的下表面为止的值。关于下部包层形成用树脂膜的厚度,并无特别限制,以硬化后的下部包层4的厚度变成所述范围的方式调整厚度。芯部2的高度并无特别限制,但优选10μm~150μm。若芯部的高度为10μm以上,则在形成光导波管后的与光收发元件或光纤的结合中,对准公差不会变小,若为150μm以下,则在形成光导波管后的与光收发元件或光纤的结合中,结合效率不会变小。就以上的观点而言,芯部的高度更优选15μm~130μm,特优选20μm~120μm。再者,关于芯部形成用树脂膜的厚度,并无特别限制,以硬化后的芯部的高度变成所述范围的方式调整厚度。上部包层3的厚度只要是可埋入芯部2的范围,则并无特别限制,但优选以干燥后的厚度计为12μm~500μm。作为上部包层3的厚度,可与最初形成的下部包层4的厚度相同,也可不同,但就埋入芯部2这一观点而言,优选比下部包层4的厚度厚。再者,所谓上部包层3的厚度,是指自芯部2与下部包层4的边界至上部包层3的上表面为止的值。在本发明的光导波管中,波长850nm的光源中的光传播损耗优选0.3db/cm以下,更优选0.2db/cm以下,特优选0.1db/cm以下。另外,实施温度85℃、湿度85%的高温高湿放置试验1000小时后的波长850nm的光源中的光传播损耗优选0.3db/cm以下,更优选0.2db/cm以下,特优选0.1db/cm以下。再者,所述高温高湿放置试验是指在依据日本电子封装与电路协会(japanelectronicspackagingcircuitsassociation,jpca)规格(jpca-pe02-05-01s)的条件下实施的高温高湿放置试验。进而,实施温度-55℃与125℃之间的温度循环试验1000次后的波长850nm的光源中的光传播损耗优选0.3db/cm以下,更优选0.2db/cm以下,特优选0.1db/cm以下。再者,所述温度循环试验是指在依据jpca规格(jpcape02-05-01s)的条件下实施的温度循环试验。另外,实施最高温度为265℃的回流试验3次后的波长850nm的光源中的光传播损耗优选0.3db/cm以下,更优选0.2db/cm以下,特优选0.1db/cm以下。再者,所述回流试验是指在依据美国电子工程设计发展联合协会(jointelectrondeviceengineeringcouncil,jedec)规格(jedecjesd22a113e)的条件下实施的无铅再流焊试验。本发明的光导波管的透明性、环境可靠性、及耐热性优异,可用作光模块的光传输管。作为光模块的形态,例如可列举:将光纤连接于光导波管的两端而成的带有光纤的光导波管、将连接器连接于光导波管的两端而成的带有连接器的光导波管、将光导波管与印刷配线板复合化而成的光电复合基板、将光导波管与使光信号与电信号相互转换的光/电转换元件组合而成的光电转换模块、将光导波管与波长分割滤光片组合而成的波长复用器/解复用器(wavelengthmultiplexer/demultiplexer)等。再者,在光电复合基板中,作为进行复合化的印刷配线板,并无特别限制,例如可列举:玻璃环氧基板、陶瓷基板等刚性基板;聚酰亚胺基板、聚对苯二甲酸乙二酯基板等柔性基板等。以下,对用以使用本发明的光导波管形成用的树脂组合物或光导波管形成用树脂膜来形成光导波管的制造方法进行说明。作为制造光导波管的方法,并无特别限制,例如可列举:使用所述树脂组合物或膜,在基材上形成光导波管形成用树脂层来制造的方法等。作为形成光导波管形成用树脂层的方法,并无特别限制,例如可列举:使用光导波管形成用树脂组合物,利用旋涂法、浸涂法、喷雾法、棒涂法、辊涂法、帘涂法、凹版涂布法、网版涂布法、喷墨涂布法等来进行涂布的方法等。当光导波管形成用树脂组合物经所述有机溶剂稀释而变成光导波管形成用树脂清漆时,视需要可在形成树脂层后,加入进行干燥的步骤。作为干燥方法,并无特别限制,例如可列举:加热干燥、减压干燥等。另外,视需要也可将这些并用。作为形成光导波管形成用树脂层的其他方法,可列举使用所述光导波管形成用树脂膜,利用层叠法来形成的方法。这些之中,就能够形成平坦性优异、具有线宽或间隙小的微细图案的光导波管这一观点而言,优选使用光导波管形成用树脂膜并利用层叠法来制造的方法。继而,对用以将光导波管形成用树脂膜用于下部包层、芯部、及上部包层来形成光导波管1的制造方法进行说明,但本发明并不受所述制造方法任何限制。首先,作为第1步骤,将下部包层形成用树脂膜层叠于基材5上。作为第1步骤中的层叠方法,并无特别限制,例如可列举:使用辊式层压机或平板型层压机,一面进行加热一面进行压接,由此进行层叠的方法等。再者,本发明中的平板型层压机是指通过将层叠材料夹在一对平板之间,并对平板加压来进行压接的层压机,例如可适宜地使用真空加压式层压机。层压温度并无特别限制,但优选20℃~130℃,层压压力并无特别限制,但优选0.1mpa~1.0mpa。当在下部包层形成用树脂膜中存在保护膜时,在去除保护膜后进行层叠。当使用真空加压式层压机进行层叠时,可使用辊式层压机,事先将下部包层形成用树脂膜临时贴附于基材5上。此处,就提升密接性及追随性的观点而言,优选一面进行压接一面进行临时贴附,在进行压接时,可使用具有热辊的层压机,一面进行加热一面进行压接。层压温度优选20℃~150℃,更优选40℃~130℃。若为所述范围,则树脂膜与基材的密接性提升,树脂层在辊层压时不会过度流动,可获得所需的膜厚。层压压力并无特别限制,但优选0.2mpa~0.9mpa,层压速度并无特别限制,但优选0.1m/min~3m/min。利用光和/或热来使层叠于基材5上的下部包层形成用树脂层硬化,而形成下部包层4。再者,下部包层形成用树脂膜的支撑膜的去除可在硬化前及硬化后的任一时期进行。利用光来使下部包层形成用树脂层硬化时的光化射线的照射量并无特别限制,但优选设为0.1j/cm2~5j/cm2。另外,当光化射线透过基材时,为了有效地进行硬化,可使用能够自两面同时照射光化射线的双面曝光机。另外,也可一面进行加热一面照射光化射线。再者,作为光硬化前后的处理,视需要可进行50℃~200℃的加热处理。利用热来使下部包层形成用树脂层硬化时的加热温度并无特别限制,但优选设为50℃~200℃。当使下部包层形成用树脂膜的支撑膜作为光导波管1的保护膜5发挥功能时,可不层叠下部包层形成用树脂膜,利用光和/或热以与所述相同的条件进行硬化,而形成下部包层4。再者,下部包层形成用树脂膜的保护膜可在硬化前去除,也可在硬化后去除。作为第2步骤,以与第1步骤相同的方法在下部包层4上层叠芯部形成用树脂膜。此处,芯部形成用树脂层优选的是以折射率高于下部包层形成用树脂层的方式设计、且包含可利用光化射线而形成芯部2(芯图案)的感光性树脂组合物。作为第3步骤,对芯部2进行曝光。作为对芯部2进行曝光的方法,并无特别限制,例如可列举:透过被称为工艺图(artwork)的负型光掩模呈图像状地照射光化射线的方法、不透过负型光掩模而使用激光直接描绘将光化射线直接照射在图像上的方法等。作为光化射线的光源,并无特别限制,例如可列举:超高压水银灯、高压水银灯、水银蒸气弧光灯、金属卤化物灯、氙气灯、碳弧灯等有效地放射紫外线的光源;照相用泛光灯、日光灯等有效地放射可见光线的光源等。对芯部2进行曝光时的光化射线的照射量优选0.01j/cm2~10j/cm2,更优选0.03j/cm2~5j/cm2,进而更优选0.05j/cm2~3j/cm2。若为所述范围,则硬化反应充分地进行,芯部不会因显影而流失,另一方面,芯部不会因曝光量过多而变粗,可形成微细的图案而适宜。芯部2的曝光可隔着芯部形成用树脂膜的支撑膜来进行,也可在去除支撑膜后进行。另外,在曝光后,就提升芯部2的分辨率及密接性的观点而言,视需要可进行曝光后加热。自紫外线照射至曝光后加热为止的时间优选10分钟以内,但所述条件并无特别限制。曝光后加热温度优选40℃~160℃,时间优选30秒~10分钟,但这些条件并无特别限制。作为第4步骤,在隔着芯部形成用树脂膜的支撑膜进行曝光的情况下,将所述支撑膜去除,并使用显影液进行显影。作为显影方法,并无特别限制,例如可列举:喷雾法、浸渍法、覆液法、旋转法、刷洗法、刮削法等。另外,视需要也可并用这些显影方法。作为显影液,并无特别限制,例如可列举:有机溶剂、包含有机溶剂与水的准水系显影液、包含碱性水溶液的水系碱性显影液、包含碱性水溶液与有机溶剂的准水系碱性显影液等。显影温度结合芯部形成用树脂层的显影性来调节。作为有机溶剂,并无特别限制,例如可适宜地列举与用于所述光导波管形成用树脂组合物的稀释的有机溶剂相同者。这些化合物可单独使用、或将两种以上组合使用。另外,在有机溶剂中也可混入表面活性剂、消泡剂等。作为准水系显影液,只要是包含一种以上的有机溶剂与水者,则并无特别限制。有机溶剂的浓度优选5质量%~90质量%。另外,在准水系显影液中也可混入少量的表面活性剂、消泡剂等。作为碱性水溶液的碱,并无特别限制,例如可列举:氢氧化锂、氢氧化钠、氢氧化钾等碱金属氢氧化物;碳酸锂、碳酸钠、碳酸钾等碱金属碳酸盐;碳酸氢锂、碳酸氢钠、碳酸氢钾等碱金属碳酸氢盐;磷酸钾、磷酸钠等碱金属磷酸盐;焦磷酸钠、焦磷酸钾等碱金属焦磷酸盐;四硼酸钠、偏硅酸钠等钠盐;碳酸铵、碳酸氢铵等铵盐;氢氧化四甲基铵、三乙醇胺、乙二胺、二乙三胺、2-氨基-2-羟基甲基-1,3-丙二醇、1,3-二氨基丙醇-2-吗啉等有机碱等。这些化合物可单独使用、或将两种以上组合使用。水系碱性显影液的ph优选9~14。另外,在水系碱性显影液中也可混入表面活性剂、消泡剂等。作为准水系碱性显影液,只要是包含碱性水溶液与一种以上的所述有机溶剂者,则并无特别限制。再者,作为有机溶剂,并无特别限制,例如可适宜地列举与用于所述光导波管形成用树脂组合物的稀释的有机溶剂相同者。优选在可充分地进行显影的范围内,尽可能减小准水系碱性显影液的ph,优选ph为8~13,更优选ph为9~12。有机溶剂的浓度通常优选5质量%~90质量%。另外,在准水系碱性显影液中也可混入少量的表面活性剂、消泡剂等。作为显影后的处理,视需要可使用有机溶剂、准水系清洗液、或水进行清洗。作为清洗方法,并无特别限制,例如可列举:喷雾法、浸渍法、覆液法、旋转法、刷洗法、刮削法等。另外,视需要也可并用这些清洗方法。有机溶剂可单独使用、或将两种以上组合使用。在准水系清洗液中,有机溶剂的浓度通常优选设为5质量%~90质量%。另外,清洗温度结合芯部形成用树脂层的显影性来调节。作为显影或清洗后的处理,就提升芯部2的硬化性及密接性的观点而言,视需要可进行曝光和/或加热。加热温度并无特别限制,但优选40℃~200℃,光化射线的照射量并无特别限制,但优选0.01j/cm2~10j/cm2。作为第5步骤,以与第1步骤及第2步骤相同的方法在下部包层4及芯部2上层叠上部包层形成用树脂膜。此处,上部包层形成用树脂层以折射率低于芯部形成用树脂层的方式设计。另外,上部包覆形成用树脂层的厚度优选大于芯部2的高度。以与第1步骤相同的方法利用光和/或热来使上部包层形成用树脂层硬化,而形成上部包层3。利用光来使上部包层形成用树脂层硬化时的光化射线的照射量并无特别限制,但优选设为0.1j/cm2~30j/cm2。另外,当光化射线透过基材时,为了有效地进行硬化,可使用能够自两面同时照射光化射线的双面曝光机。另外,视需要可一面进行加热一面照射光化射线,作为光硬化前后的处理,可进行加热处理。光化射线照射过程中和/或照射后的加热温度并无特别限制,但优选50℃~200℃。利用热来使上部包层形成用树脂层硬化时的加热温度并无特别限制,但优选50℃~200℃。再者,当需要去除上部包层形成用树脂膜的支撑膜时,可在硬化前去除,也可在硬化后去除。可利用以上的步骤来制作光导波管1。[实施例]以下,通过实施例来对本发明进行具体说明,但本发明并不限定于这些实施例。再者,各例中的份均为重量份,物性的测定利用以下所示的方法来进行。1)共聚合物(可溶性多官能芳香族共聚合物)的分子量及分子量分布分子量及分子量分布测定是使用凝胶渗透色谱仪(gelpermeationchromatograph,gpc)(东曹制造,hlc-8120gpc),将四氢呋喃用于溶媒,以流量1.0ml/min、管柱温度38℃,使用利用单分散聚苯乙烯的校准曲线来进行。2)共聚合物的结构使用日本电子制造的jnm-la600型核磁共振分光装置,利用13c-核磁共振(nuclearmagneticresonance,nmr)及1h-nmr分析来决定。使用氯仿-d1作为溶媒,并使用四甲基硅烷的共振线作为内部标准。3)末端基的分析末端基的算出是根据自所述gpc测定所获得的数量平均分子量、与自1h-nmr测定及气相色谱仪(gaschromatograph,gc)分析的结果所获得的相对于单体总量的用于导入末端基的衍生物量,算出具有末端基的可溶性多官能乙烯基芳香族共聚合物1分子中所含有的末端基数。4)硬化物的玻璃化温度(tg)及软化温度测定以干燥后的厚度变成20μm的方式,将共聚合物溶液均匀地涂布于玻璃基板上,使用热板在90℃下加热30分钟来进行干燥。将玻璃基板与所获得的树脂膜一同设置于热机械分析仪(thermomechanicalanalyzer,tma)(热机械分析装置)上,在氮气气流下,以升温速度10℃/min升温至220℃为止,进而在220℃下进行20分钟加热处理,由此去除残存的溶媒,同时使共聚合物硬化。将玻璃基板放置冷却至室温为止后,使分析用探针接触tma测定装置中的试样,在氮气气流下,以升温速度10℃/min自30℃至360℃为止进行扫描测定,并利用切线法来求出软化温度。关于玻璃化温度,将所述试验片设置于动态机械分析仪(dynamicmechanicalanalyzer,dma)(动态粘弹性装置)测定装置上,在氮气气流下,以升温速度3℃/min自30℃至320℃为止进行扫描,由此进行测定,并利用tanδ曲线的峰顶来求出tg。5)耐热性评价及耐热变色性的测定共聚合物的耐热性评价是将试样设置于热重分析仪(thermalgravimetricanalyzer,tga)(热天平)测定装置上,在氮气气流下,以升温速度10℃/min自30℃至400℃为止进行扫描,由此进行测定,并求出350℃下的重量减少作为耐热性。另一方面,耐热变色性的测定是将共聚合物6.0g、甲基丙烯酸苄酯4.0g、及过氧化-2-乙基己酸叔丁酯0.02g混合,在氮气气流下以200℃加热1小时,而获得硬化物。然后,以目视确认所获得的硬化物的变色量,并分类成○:无热变色,δ:淡黄色,×:黄色,由此进行耐热变色性的评价。6)相容性的测定共聚合物与环氧树脂的相容性的测定是使试样5.0g、环氧树脂(液状双酚a型环氧树脂:日本环氧树脂公司制造,艾比克特(epikote)828)3.0g、及酚树脂(三聚氰胺骨架系酚树脂:群荣化学工业公司制造,ps-6492)2.0g溶解于甲基乙基酮(methylethylketone,mek)10g中,以目视确认溶解后的试样的透明性,并分类成○:透明,δ:半透明,×:不透明或未溶解,由此进行相容性的评价。合成例1将二乙烯基苯(1,4-二乙烯基苯及1,3-二乙烯基苯的混合物,以下的例子也相同)1.28摩尔(182.7ml)、乙基乙烯基苯(1-乙基-4-乙烯基苯及1-乙基-3-乙烯基苯的混合物,以下的例子也相同)0.97摩尔(137.8ml)、甲基丙烯酸叔丁酯2.00摩尔(323.2ml)、甲苯300ml投入至2.0l的反应器内,在50℃下添加50毫摩尔的三氟化硼的二乙基醚络合物,并进行4小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在60℃下减压脱挥,并回收聚合体。称量所获得的聚合体,确认获得234.6g的共聚合物a。所获得的共聚合物a的mn为842,mw为3640,mw/mn为4.32。通过进行13c-nmr及1h-nmr分析,共聚合物a观察到源自甲基丙烯酸叔丁酯的末端基的共振线。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自甲基丙烯酸叔丁酯的结构单元的导入量(c1)为2.3(个/分子)。另外,含有60.1摩尔%的源自二乙烯基苯的结构单元及合计39.9摩尔%的源自乙基乙烯基苯的结构单元(末端结构单元除外)。共聚合物a中所含有的乙烯基含量为36.2摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为2.80wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为○。共聚合物a可溶于甲苯、二甲苯、四氢呋喃(tetrahydrofuran,thf)、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。合成例2将二乙烯基苯1.28摩尔(182.7ml)、乙基乙烯基苯0.97摩尔(137.8ml)、丙烯酸叔丁酯2.00摩尔(289.6ml)、甲苯300ml投入至2.0l的反应器内,在50℃下添加50毫摩尔的三氟化硼的二乙基醚络合物,并进行4小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在60℃下减压脱挥,并回收聚合体。称量所获得的聚合体,确认获得215.3g的共聚合物b。所获得的共聚合物b的mn为952,mw为4490,mw/mn为4.72。通过进行13c-nmr及1h-nmr分析,共聚合物b观察到源自丙烯酸叔丁酯的末端基的共振线。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自丙烯酸叔丁酯的结构单元的导入量(c1)为2.5(个/分子)。另外,含有59.3摩尔%的源自二乙烯基苯的结构单元及合计40.7摩尔%的源自乙基乙烯基苯的结构单元(末端结构单元除外)。共聚合物b中所含有的乙烯基含量为34.7摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为3.04wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为○。共聚合物b可溶于甲苯、二甲苯、thf、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。合成例3(比较例)将二乙烯基苯2.03摩尔(288.5ml)、乙基乙烯基苯0.084摩尔(12.0ml)、苯乙烯2.11摩尔(241.7ml)、甲基丙烯酸2-苯氧基乙酯2.25摩尔(427.3ml)、乙酸丁酯100.0ml、甲苯1150ml投入至3.0l的反应器内,在50℃下添加300毫摩尔的三氟化硼的二乙基醚络合物,并进行4小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在室温下将反应混合液投入至大量的甲醇中,使聚合体析出。利用甲醇对所获得的聚合体进行清洗,并进行滤除、干燥、称量,而获得282.4g的共聚合物c。所获得的共聚合物c的mn为2030,mw为5180,mw/mn为2.55。通过进行13c-nmr及1h-nmr分析,共聚合物c观察到源自甲基丙烯酸2-苯氧基乙酯的末端基的共振线。进行共聚合物c的元素分析的结果,c:87.3wt%,h:7.4wt%,o:5.2wt%。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自甲基丙烯酸2-苯氧基乙酯的结构单元的导入量(c1)为2.3(个/分子)。另外,含有59.2摩尔%的源自二乙烯基苯的结构单元及合计40.8摩尔%的源自苯乙烯与乙基苯的结构单元(末端结构单元除外)。共聚合物c中所含有的乙烯基含量为35.3摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为4.86wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为δ。共聚合物c可溶于甲苯、二甲苯、thf、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。合成例4(比较例)将二乙烯基苯1.92摩尔(273.5ml)、乙基乙烯基苯0.08摩尔(11.4ml)、苯乙烯2.0摩尔(229.2ml)、丙烯酸2-苯氧基乙酯2.00摩尔(348.1ml)、乙酸丁酯250.0ml、甲苯1000ml投入至3.0l的反应器内,在70℃下添加80毫摩尔的三氟化硼的二乙基醚络合物,并进行6小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在室温下将反应混合液投入至大量的甲醇中,使聚合体析出。利用甲醇对所获得的聚合体进行清洗,并进行滤除、干燥、称量,而获得164.2g的共聚合物d。所获得的共聚合物d的mn为2330,mw为4940,mw/mn为2.12。通过进行13c-nmr及1h-nmr分析,共聚合物d观察到源自丙烯酸2-苯氧基乙酯的末端基的共振线。进行共聚合物d的元素分析的结果,c:84.4wt%,h:7.3wt%,o:7.9wt%。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自丙烯酸2-苯氧基乙酯的结构单元的导入量(c1)为3.5(个/分子)。另外,含有54.3摩尔%的源自二乙烯基苯的结构单元及合计45.7摩尔%的源自苯乙烯与乙基苯的结构单元(末端结构单元除外)。共聚合物d中所含有的乙烯基含量为21.8摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为5.50wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为δ。共聚合物d可溶于甲苯、二甲苯、thf、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。合成例5将二乙烯基苯1.024摩尔(146.2ml)、乙基乙烯基苯0.776摩尔(110.2ml)、苯乙烯0.45摩尔(51.7ml)、甲基丙烯酸叔丁酯2.00摩尔(323.2ml)、甲苯300ml投入至2.0l的反应器内,在50℃下添加50毫摩尔的三氟化硼的二乙基醚络合物,并进行4小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在60℃下减压脱挥,并回收聚合体。称量所获得的聚合体,确认获得225.7g的共聚合物e。所获得的共聚合物e的mn为789,mw为3450,mw/mn为4.37。通过进行13c-nmr及1h-nmr分析,共聚合物e观察到源自甲基丙烯酸叔丁酯的末端基的共振线。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自甲基丙烯酸叔丁酯的结构单元的导入量(c1)为2.4(个/分子)。另外,含有47.1摩尔%的源自二乙烯基苯的结构单元、34.0摩尔%的源自乙基乙烯基苯的结构单元、及18.9摩尔%的源自苯乙烯的结构单元(末端结构单元除外)。共聚合物e中所含有的乙烯基含量为33.8摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为3.37wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为○。共聚合物e可溶于甲苯、二甲苯、thf、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。合成例6将二乙烯基苯1.024摩尔(146.2ml)、乙基乙烯基苯0.776摩尔(110.2ml)、二乙烯基联苯0.45摩尔(81.1g)、甲基丙烯酸叔丁酯2.00摩尔(323.2ml)、甲苯300ml投入至2.0l的反应器内,在50℃下添加50毫摩尔的三氟化硼的二乙基醚络合物,并进行4小时反应。利用碳酸氢钠水溶液来使聚合停止后,利用纯水将油层清洗3次,在60℃下减压脱挥,并回收聚合体。称量所获得的聚合体,确认获得318.7g的共聚合物f。所获得的共聚合物f的mn为913,mw为4210,mw/mn为4.61。通过进行13c-nmr及1h-nmr分析,共聚合物f观察到源自甲基丙烯酸叔丁酯的末端基的共振线。根据元素分析结果与标准聚苯乙烯换算的数量平均分子量所算出的可溶性多官能乙烯基芳香族聚合体的源自甲基丙烯酸叔丁酯的结构单元的导入量(c1)为2.2(个/分子)。另外,含有47.3摩尔%的源自二乙烯基苯的结构单元、34.5摩尔%的源自乙基乙烯基苯的结构单元、及18.2摩尔%的源自苯乙烯的结构单元(末端结构单元除外)。共聚合物f中所含有的乙烯基含量为32.9摩尔%(末端结构单元除外)。另外,硬化物的tma测定的结果,未观察到明确的tg,软化温度为300℃以上。tga测定的结果,350℃下的重量减少为2.92wt%,耐热变色性为○。另一方面,与环氧树脂的相容性为○。共聚合物f可溶于甲苯、二甲苯、thf、二氯乙烷、二氯甲烷、氯仿中,未看见凝胶的生成。使用合成例1~合成例6中所获得的共聚合物a~共聚合物f,根据下述的试验方法进行使用这些树脂的硬化性树脂组合物的清漆、及硬化物的特性的评价。7)溶液粘度硬化性树脂组合物的溶液粘度是使用e型粘度计,在测定温度:25℃下进行测定。8)弯曲强度及弯曲断裂伸长率用于弯曲试验的试验片是将硬化性树脂组合物的清漆装载于真空压制成形机的下方的模具上,并在加热真空下使溶剂脱挥。其后,装载上模,在真空下进行加热压制,并在200℃下保持1小时,由此使厚度:1.0mm的平板成形。自进行成形所获得的平板制作宽度:5.0mm、厚度:1.0mm、长度:120mm的试验片,并进行弯曲试验。所制作的弯曲试验片的弯曲强度及弯曲断裂伸长率是使用万能试验装置进行测定。而且,弯曲强度及弯曲断裂伸长率是将相对于成为基准的调配的测定值变成未满±10%的值者评价为○,将相对于成为基准的调配的测定值变成10%以上的值者评价为◎,将相对于成为基准的调配的测定值变成-10%~-20%的范围的值者评价为δ,将相对于成为基准的调配的测定值变成-20%以下的值者评价为×。9)线膨胀系数及玻璃化温度用于硬化性树脂组合物的线膨胀系数及玻璃化温度的试验的试验片是将硬化性树脂组合物的清漆装载于真空压制成形机的下方的平板形状的模具上,并在加热真空下使溶剂脱挥。其后,隔着0.2mm的间隔物装载上模,在真空下进行加热压制,并在200℃下保持1小时,由此使厚度:0.2mm的平板成形。自进行成形所获得的平板制作宽度:3.0mm、厚度:0.2mm、长度:40mm的试验片,并仅设置于tma(热机械分析装置)的上方的夹头上,在氮气气流下,以升温速度10℃/min升温至220℃为止,进而在220℃下进行20分钟加热处理,由此去除残存的溶媒,并且进行试验片中的成形应变的去除。将tma放置冷却至室温为止后,将tma测定装置中的试验片的下侧也设置于分析用探针上,在氮气气流下,以升温速度10℃/min自30℃至360℃为止进行扫描测定,并根据0℃~40℃中的尺寸变化来算出线膨胀系数。另外,关于玻璃化温度,将所述试验片设置于dma(动态粘弹性装置)测定装置上,在氮气气流下,以升温速度3℃/min自30℃至320℃为止进行扫描,由此进行测定,并利用tanδ曲线的峰顶来求出tg。10)介电常数及介电损耗正切依据日本工业标准(japanindustrialstandards,jis)c2565规格,利用aet股份有限公司制造的空腔谐振器法介电常数测定装置,并使用绝对干燥后在23℃、湿度50%的室内保管24小时后的硬化物平板试验片,测定18ghz中的介电常数及介电损耗正切。另外,将硬化物平板试验片在85℃、相对湿度85%中放置2周后,进行介电常数及介电损耗正切的测定,并测定耐湿热试验后的介电常数及介电损耗正切。11)铜箔剥离强度将玻璃布(e玻璃,单位面积重量为71g/m2)浸渍于热硬化性树脂组合物的清漆中来进行含浸,并在80℃的空气烘箱中进行10分钟干燥。此时,以所获得的预浸料的树脂含量(resincontent,r.c)变成50wt%的方式进行调整。使用所述预浸料,以成形后的厚度变成约0.6mm~1.0mm的方式,视需要将多片所述硬化性复合材料叠加,在其两面放置厚度为18μm的铜箔(商品名为f2-ws铜箔,rz:2.0μm,ra:0.3μm)并利用真空压制成形机来进行成形硬化而获得评价用层叠体。硬化条件为以3℃/min升温,并在压力3mpa下以200℃保持60分钟,而获得评价用覆铜层叠板。自以所述方式获得的层叠体硬化物切出宽度为20mm、长度为100mm的试验片,在铜箔面切入宽度为10mm的平行的切口后,在相对于面为90°的方向上以50mm/min的速度连续地剥离铜箔,利用拉伸试验机测定此时的应力,将其应力的最低值记录为铜箔剥离强度。(依据jisc6481)。耐湿热性试验后的铜箔剥离强度的试验是将所述试验片在85℃、相对湿度85%中放置2周后,以与所述相同的方式进行测定。12)镀铜剥离强度·带有镀铜的层叠板的制作在40℃下,使前项所制作的覆铜层叠板在过硫酸铵150g/l的水溶液中浸渍20分钟来对铜箔进行蚀刻去除。继而,利用浸渍法,在80℃下,在膨润水溶液的萨库珀吉mlb调节剂(circupositmlbconditioner)211(日本罗门哈斯(rohmandhaasjapan)股份有限公司制造,商品名)中对未抽出试样的层叠板进行5分钟浸渍处理。进而,在流水洗的室温下进行3分钟处理后,使用萨库珀吉mlb促进剂(circupositmlbpromoter)213(日本罗门哈斯股份有限公司制造,商品名)作为高锰酸强碱水溶液,同样地利用浸渍法在80℃下进行10分钟浸渍处理。继而,使用mlb中和剂(mlbneutralizer)216(日本罗门哈斯股份有限公司制造,商品名)作为中和液,利用浸渍法在40℃下进行5分钟浸渍处理。在流水洗的室温下进行3分钟处理后,使用调节液的clc-501(商品名,日立化成工业股份有限公司制造)在60℃下进行5分钟处理,并进行流水洗,在预浸液pd-201(商品名,日立化成工业股份有限公司制造)水溶液中,在室温下进行3分钟处理,然后在含有金属钯液hs-202b(商品名,日立化成工业股份有限公司制造)的水溶液中,在室温下进行10分钟处理,并进行水洗,然后在活化处理液adp-501(商品名,日立化成工业股份有限公司制造)水溶液中,在室温下进行5分钟处理。而且,使用卡斯特(cust)-201作为无电解镀铜液,利用浸渍法在室温下进行15分钟浸渍处理,由此在层叠板的两面形成无电解铜厚为0.5μm的基底铜,进而利用电解铜镀敷加厚至铜厚为20μm为止。而且,自所述带有镀层的试验用层叠板硬化物,利用蚀刻来加工成铜宽为10mm、长度为100mm的线,将其一端剥下并利用夹具夹持,依据jis-c-6421,将在室温中在垂直方向上剥离约50mm时的负荷的最低值记录为镀铜剥离强度。13)成形性使用前项中进行了成形的评价用覆铜层叠板,呈格子状地制作图案化成线宽(l)为0.5mm、线间隔(s)为1.0mm(l/s=0.5/1.0mm)的芯材。对所述芯材进行黑化处理,继而,在其上进而层叠预浸料,进行2次成形,由此制作内层为格子状图案的评价用层叠基板。针对所制作的评价用层叠基板,例如确认是否产生由树脂清漆的流动性不足所形成的空隙等缺陷。其后,将所述评价用层叠基板在沸水中浸渍4小时后,浸渍于280℃的焊料槽中。此时,将无法确认空隙的存在,浸渍于焊料槽中后也未看到膨胀、层间剥离、白点(白斑)等不良现象的产生者评价为“○”,将可确认空隙、膨胀、层间剥离、白点(白斑)的任一者的产生者评价为“×”。<表中的记号的说明>改性ppe-a:两末端具有乙烯基的聚亚苯基寡聚物(mn=1160,三菱瓦斯化学(股份)制造,2,2',3,3',5,5'-六甲基联苯-4,4'-二醇·2,6-二甲基苯酚缩聚物与氯甲基苯乙烯的反应产物)改性ppe-b:两末端具有乙烯基的聚亚苯基寡聚物(mn=2270,三菱瓦斯化学(股份)制造,2,2',3,3',5,5'-六甲基联苯-4,4'-二醇·2,6-二甲基苯酚缩聚物与氯甲基苯乙烯的反应产物)改性ppe-c:一末端具有乙烯基的聚亚苯基寡聚物(mn=2340,聚苯醚(沙伯基础创新塑料(sabicinnovativeplastics)公司制造的sa120)与氯甲基苯乙烯的反应产物)taic:三烯丙基异氰脲酸酯(日本化成股份有限公司制造)dcp:三环癸烷二甲醇二甲基丙烯酸酯(新中村化学工业股份有限公司制造)a-dcp:三环癸烷二甲醇二丙烯酸酯(新中村化学工业股份有限公司制造)邻甲酚酚醛清漆型环氧树脂:艾博托特(epotohto)ydcn-700-3(低粘度型,新日铁住金化学股份有限公司制造)双酚f型液状环氧树脂:艾比克特(epikote)806l,mw=370(日本环氧树脂公司制造)萘骨架液状环氧树脂:艾比克隆(epiclon)hp-4032d,mw=304(迪爱生公司制造)萘酚型环氧树脂:esn-475v,环氧当量:340(新日铁住金化学公司制造)双酚a型液状环氧树脂:艾比克特(epikote)828us,mw=370(日本环氧树脂公司制造)改性ppe-d:两末端具有环氧基的聚亚苯基寡聚物(mn=1180,三菱瓦斯化学(股份)制造,2,2',3,3',5,5'-六甲基联苯-4,4'-二醇·2,6-二甲基苯酚缩聚物与表氯醇的反应产物)联苯骨架酚树脂:明和化成公司制造,meh-7851-s三聚氰胺骨架系酚树脂:群荣化学工业公司制造,ps-6492含有烯丙基的骨架酚树脂:日本环氧树脂公司制造,ylh-903脂环式骨架酸酐:新日本理化公司制造,mh-700芳香族骨架酸酐:日本沙多玛(sartomer·japan)公司制造,sma树脂(resin)ef60苯乙烯系共聚合物:科腾(kraton)a1535(科腾聚合物有限公司(kratonpolymersllc)制造)苯氧基树脂:重量平均分子量为37000,三菱化学(股份)制造的“yl7553bh30”(不挥发成分为30质量%的mek与环己酮的1:1溶液)非晶球状二氧化硅:雅都玛(admatechs)公司制造,se2050spe,平均粒径为0.5μm(利用苯基硅烷偶合剂进行处理)实施例5使合成例1中所获得的共聚合物-a20g与作为聚合引发剂的帕库米(percumyl)p0.2g、作为抗氧化剂的ao-600.2g溶解于甲苯8.6g中而获得硬化性树脂组合物(清漆a)。将所制备的清漆a滴加于下模上,在减压下以130℃使溶媒脱挥后,组合堆起模具,在200℃、3mpa的条件下进行1小时真空加压压制,而进行热硬化。针对所获得的厚度:0.2mm的硬化物平板试验片,测定以18ghz的介电常数与介电损耗正切为首的各种特性。另外,将硬化物平板试验片在85℃、相对湿度85%下放置2周后,进行介电常数及介电损耗正切的测定,并测定耐湿热试验后的介电常数及介电损耗正切。将通过这些测定所获得的结果示于表1中。[表1]实施例6~实施例8、比较例3~比较例4除设为表1中所示的调配配方以外,以与实施例5相同的方法获得硬化性树脂组合物(清漆)。而且,以与实施例5相同的方式制作硬化物平板试验片,并对与实施例5相同的项目进行试验·评价。将通过这些试验所获得的结果示于表1中。实施例9~实施例19、比较例5~比较例8除设为表2及表3中所示的调配配方以外,以与实施例5类似的方法获得硬化性树脂组合物(清漆)。而且,以与实施例5相同的方式制作硬化物平板试验片,并对与实施例5相同的项目进行试验·评价。将通过这些试验所获得的结果示于表1中。进而,使用这些实施例及比较例中所示的清漆,根据所述试验方法11)~试验方法13)中所记载的方法,制作预浸料、试验用覆铜层叠板、及试验用带有镀层的层叠板,并进行铜箔剥离强度、镀铜剥离强度、及成形性的评价。将试验结果示于表2及表3中。[表2][表3]实施例21~实施例30及比较例11~比较例12使用合成例1~合成例6中所获得的共聚合物a~共聚合物f,制成硬化性树脂组合物。以表4及表5中所示的比例(重量比)将表4及表5中所示的共聚合物、丙烯酸酯、聚合引发剂及添加剂混合,而制成硬化性树脂组合物。此处,表4及表5中所示的实施例及比较例除实施例29以外,将以表中所记载的调配进行混合所获得的树脂组合物直接成形为评价用的试验片。关于实施例29,为了使稳定剂及引发剂的溶解变得容易,相对于树脂成分100重量份,添加作为溶剂的甲苯30重量份,使各成分溶解而制成树脂清漆,在即将进行成形前以50℃对树脂清漆进行减压脱挥,使用去除溶剂后的树脂组合物来制成试验片。试验片在仅添加热引发剂的调配(实施例21~实施例28、比较例11~比较例12)中,将硬化性树脂组合物装载于下模上,在加热真空下进行b-阶段化。树脂组合物的粘度上升而变得黏稠后,将上模装载于下模上,并将模具装载于真空压制成形机的热板上,在真空下进行加热压制,并在200℃下保持1小时,由此使厚度:1.0mm的硬化树脂平板a1成形。另一方面,在含有紫外线(ultraviolet,uv)引发剂的调配(实施例29~实施例30)中,在宽度为50mm、长度为50mm、厚度为1.0mm的2片玻璃板之间,使用厚度为1.0mm的硅橡胶制的间隔物,空开1.0mm厚的间隙向利用聚酰亚胺带将外周卷绕固定的玻璃模具中注入组合物,利用高压水银灯自所述玻璃模具的一面照射几秒紫外线来进行1次硬化后,将所述玻璃模具放入至氮气气流下的惰性气体烘箱中,在200℃下加热1小时,由此使硬化树脂平板a2成形。将使厚度为1.0mm的硬化树脂平板a1或硬化树脂平板a2变成规定的大小所获得的试验片设为试验片a。将使以与所述相同的方式获得的厚度为0.2mm的硬化树脂平板b1或硬化树脂平板b2变成规定的大小所获得的试验片设为试验片b。根据下述的试验方法进行这些硬化性树脂组合物、及硬化物的特性的评价。将这些的测定结果示于表4及表5中。7)溶液粘度硬化性树脂组合物(不含溶剂)的溶液粘度是使用e型粘度计,在测定温度:25℃下进行测定。8)弯曲强度及弯曲断裂伸长率准备宽度:5.0mm、厚度:1.0mm、长度:120mm的试验片a。弯曲强度及弯曲断裂伸长率是使用万能试验装置进行测定。而且,弯曲强度及弯曲断裂伸长率是将相对于成为基准的调配的测定值变成未满±10%的值者评价为○,将相对于成为基准的调配的测定值变成10%以上的值者评价为◎,将相对于成为基准的调配的测定值变成-10%~-20%的范围的值者评价为δ,将相对于成为基准的调配的测定值变成-20%以下的值者评价为×。9)线膨胀系数及玻璃化温度准备宽度:3.0mm、厚度:0.2mm、长度:40mm的试验片b。仅设置于tma(热机械分析装置)的上方的夹头上,在氮气气流下,以升温速度10℃/min升温至220℃为止,进而在220℃下进行20分钟加热处理,由此进行试验片中的成形应变的去除。将tma放置冷却至室温为止后,将tma测定装置中的试验片的下侧也设置于分析用探针上,在氮气气流下,以升温速度10℃/min自30℃至360℃为止进行扫描测定,并根据0℃~40℃中的尺寸变化来算出线膨胀系数。另外,利用切线法来求出玻璃化温度。再者,在所有实施例及比较例中未观察到玻璃化温度,因此省略记载。10)折射率的测定使用厚度为1.0mm的试验片a,利用阿贝折射率计(爱拓(atago)(股份)制造)来测定折射率及阿贝数。12)色相使用厚度为1.0mm的试验片a,利用色彩色差计(商品名“型号tc-8600”,东京电色(股份)制造)来进行测定,并表示其黄色指数(yellownessindex,yi)值。13)雾度(浊度)及全光线透过率使用厚度为0.2mm的试验片b,利用积分球式光线透过率测定装置(日本电色公司制造,sz-σ90)测定雾度(浊度)与全光线透过率。14)脱模性、模再现性、及毛刺、漏出在仅添加热引发剂的调配(实施例21~实施例28、比较例11~比较例12)中,将硬化性树脂组合物装载于下模上,在加热真空下进行b-阶段化。树脂组合物的粘度上升而变得黏稠后,将上模装载于下模上,并将模具装载于真空压制成形机的热板上,在真空下进行加热压制,并在200℃下保持1小时,另一方面,在含有uv引发剂的调配(实施例29~实施例30)中,将硬化性树脂组合物装载于具有直径3.0mm的球面透镜的形状的模腔的模具上,并将玻璃制上模装载于下模上,利用高压水银灯自所述玻璃制上模的上表面照射几秒紫外线来进行1次硬化后,将所述玻璃模具放入至氮气气流下的惰性气体烘箱中,在200℃下加热1小时,由此使直径:3.0mm的球面透镜成形。将所述球面透镜的成形重复5次,利用使硬化树脂透镜自模具中脱模时的难易度来评价脱模性。○····自模具的脱模性良好δ····脱模略困难×····脱模困难或有模残留物模再现性是观察硬化树脂透镜的表面形状与模具的表面形状来进行评价。○····再现性良好×····再现性不良毛刺、漏出是根据使硬化树脂透镜自模具中脱模时,在成形品的制品部分以外所产生的毛刺的大小及树脂朝模具的缝隙中的漏入的程度来评价。○····毛刺的大小未满0.05mm,树脂的漏入未满1.0mmδ····毛刺的大小未满0.2mm,树脂的漏入未满3.0mm×····毛刺的大小为0.2mm以上,树脂的漏入为3.0mm以上15)回流耐热性使用厚度为1.0mm的试验片a,利用分光测色计cm-3700d(柯尼卡美能达(konicaminolta)公司制造)来测定波长:400nm的分光透过率。将测定时间点设为在190℃下进行60分钟的后硬化的耐热试验前、及在空气烘箱中以260℃进行8分钟的耐热试验后。<表中的记号的说明>方库里(fancryl)fa-bza:日立化成工业股份有限公司制造,丙烯酸苄酯方库里(fancryl)fa-302a:日立化成工业股份有限公司制造,丙烯酸邻苯基苯氧基乙酯莱特丙烯酸酯(lightacrylate)po-a:共荣社化学股份有限公司制造,丙烯酸苯氧基乙酯奥古索(ogsol)ea-0200:大阪燃气化学(osakagaschemicals)股份有限公司制造,含有芴骨架的丙烯酸酯莱特丙烯酸酯(lightacrylate)tmp-a:共荣社化学股份有限公司制造,三羟甲基丙烷三丙烯酸酯艾迪科斯塔波(adekastab)ao-412s:艾迪科(adeka)股份有限公司制造,季戊四醇-四(十二烷硫基丙酸酯)艾迪科斯塔波(adekastab)ao-60:艾迪科股份有限公司制造,季戊四醇四[3-(3,5-二-叔丁基-4-羟基苯基)丙酸酯]帕布奇(perbutyl)o:日本油脂股份有限公司制造,过氧化-2-乙基己酸叔丁酯艳佳固(irgacure)184:汽巴精化公司制造,1-羟基-环己基-苯基-酮[表4][表5]合成例7向具备搅拌机、冷却管、气体导入管、滴加漏斗、及温度计的烧瓶中添加丙二醇单甲基醚乙酸酯50重量份及乳酸甲酯19重量份,一面导入氮气一面进行搅拌。使温度上升至65℃,历时3小时滴加甲基丙烯酸甲酯45重量份、丙烯酸丁酯35重量份、甲基丙烯酸2-羟基乙酯17重量份、甲基丙烯酸13重量份、2,2'-偶氮双(2,4-二甲基戊腈)3重量份、丙二醇单甲基醚乙酸酯49重量份、及乳酸甲酯20重量份的混合物后,在65℃下搅拌3小时,进而在95℃下继续搅拌1小时,而获得共聚合物g的溶液(固体成分为45质量%)。所获得的共聚合物g的mn为16700,mw为38100,mw/mn为2.28,酸值为79mgkoh/g。制造例1一面进行搅拌一面将作为(a)成分的共聚合物a10重量份,作为(e)成分的所述共聚合物g的溶液(固体成分为45质量%)62重量份(固体成分为28质量份),作为(b)成分的具有聚酯骨架的(甲基)丙烯酸氨基甲酸酯(新中村化学工业(股份)制造的“u-200ax”)33质量份、及具有聚丙二醇骨架的(甲基)丙烯酸氨基甲酸酯(新中村化学工业(股份)制造的“ua-4200”)15质量份,作为(f)成分的利用甲基乙基酮肟保护六亚甲基二异氰酸酯的异氰脲酸酯型三聚体的多官能封闭型异氰酸酯溶液(固体成分为75质量%)(住化拜尔聚氨脂(sumikabayerurethane)(股份)制造的“斯密久(sumidur)bl3175”)20质量份(固体成分15质量份),作为(c)成分的光聚合引发剂的1-[4-(2-羟基乙氧基)苯基]-2-羟基-2-甲基-1-丙烷-1-酮(日本汽巴(ciba·japan)(股份)制造的“艳佳固(irgacure)2959”)1质量份、双(2,4,6-三甲基苯甲酰基)苯基氧化膦(日本汽巴(股份)制造的“艳佳固(irgacure)819”)1质量份,以及作为稀释用有机溶剂的丙二醇单甲基醚乙酸酯23质量份混合。使用孔径为2μm的聚四氟乙烯过滤器进行加压过滤后,进行减压脱泡,而获得包层形成用树脂清漆w-i。制造例2使用所述涂敷机,将所述清漆w-i涂布于pet膜(东洋纺(股份)制造的“克斯莫闪(cosmoshine)a4100”,厚度为50μm)的非处理面上,在100℃下进行20分钟干燥后,贴付表面脱模处理pet膜(帝人杜邦薄膜(teijindupontfilms)(股份)制造的“普雷克斯(purex)a31”,厚度为25μm)作为覆盖膜,而获得包层形成用树脂膜f-i。此时树脂层的厚度是以硬化后的膜厚在下部包层形成用树脂膜中变成20μm、及在上部包层形成用树脂膜中变成60μm的方式进行调节。制造例3调配共聚合物a15重量份、共聚合物g的溶液33.3重量份(固体成分为15重量份)、2-甲基丙烯酰氧基乙基丁二酸(共荣社化学(股份)制造的“lightesterho-ms”)10重量份、具有聚酯骨架的(甲基)丙烯酸氨基甲酸酯(新中村化学工业(股份)制造的“u-108a”)20重量份、双酚a的环氧乙烷(ethyleneoxide,eo)加合物二丙烯酸酯(日立化成工业(股份)制造的“方库里(fancryl)fa-321a”)20重量份、及多官能封闭型异氰酸酯溶液(固体成分为75重量%)(住化拜尔聚氨脂(股份)制造的“斯密久(sumidur)bl3175”)20重量份(固体成分为15重量份),进而添加1-[4-(2-羟基乙氧基)苯基]-2-羟基-2-甲基-1-丙烷-1-酮(日本巴斯夫(basfjapan)(股份)制造;艳佳固(irgacure)2959)1重量份、双(2,4,6-三甲基苯甲酰基)苯基氧化膦(日本巴斯夫(股份)制造;艳佳固(irgacure)819)1重量份、及丙二醇单甲基醚乙酸酯19重量份,一面进行搅拌一面进行混合。使用孔径为2μm的聚四氟乙烯过滤器进行加压过滤后,进行减压脱泡,而获得芯部形成用树脂清漆w-ii。制造例4使用涂敷机,将芯部形成用树脂清漆w-ii涂布于pet膜(东洋纺(股份)制造的“克斯莫闪(cosmoshine)a1517”,厚度为16μm)的非处理面上,在80℃下进行10分钟干燥,并在100℃下进行10分钟干燥后,贴付表面脱模处理pet膜(帝人杜邦薄膜(股份)制造;普雷克斯(purex)a31,厚度为25μm)作为保护膜,而获得芯部形成用树脂膜f-ii。此时树脂层的厚度是以硬化后的膜厚变成50μm的方式进行调节。实施例31使用辊式层压机,在压力为0.5mpa、温度为80℃的条件下,将去除了保护膜的下部包层形成用树脂膜f-i层叠于pet膜(厚度50μm)上。进而,使用真空加压式层压机,在压力为0.4mpa、温度为80℃的条件下进行压接。继而,使用紫外线曝光机,照射紫外线(波长365nm)2000mj/cm2后,去除支撑膜。其后,在160℃下进行1小时加热硬化,由此形成下部包层4。继而,使用辊式层压机,在压力为0.5mpa、温度为80℃的条件下,将去除了保护膜的所述芯部形成用树脂膜f-ii层叠于下部包层4上。进而,使用所述真空加压式层压机,在压力为0.4mpa、温度为80℃的条件下进行压接。继而,隔着宽度为50μm的负型光掩模,利用紫外线曝光机以1000mj/cm2照射紫外线(波长365nm),而对芯部2(芯图案)进行曝光。在80℃下进行曝光后加热后,去除支撑膜,并使用丙二醇单甲基醚乙酸酯/n,n-二甲基乙酰胺(70/30质量比)进行显影。继而,使用丙二醇单甲基醚乙酸酯进行清洗后,进而使用2-丙醇进行清洗。干燥后,在160℃下进行1小时加热硬化。继而,使用真空加压式层压机,在压力为0.4mpa、温度为100℃的条件下,将去除了保护膜的上部包层形成用树脂膜f-i层叠于芯部2及下部包层4上。以2000mj/cm2照射紫外线(波长365nm),去除支撑膜后,在160℃下进行1小时加热硬化,由此形成上部包层3。其后,去除表面脱模处理pet膜,而获得图1(d)中所示的光导波管1。其后,使用切割锯切出长度为10cm的柔性光导波管。对所获得的柔性光导波管进行以下的评价。[光传播损耗测定]将以波长850nm的光为中心波长的垂直腔面发射激光器(verticalcavitysurfaceemittinglaser,vcsel)(exfo公司制造的“fls-300-01-vcl”)用于光源,并使用光接收传感器(爱德万测试(advantest)(股份)制造的“q82214”)、入射光纤(gi-50/125多模光纤,数值孔径(numericalaperture,na)=0.20)、及出射光纤(si-114/125,na=0.22)来测定所获得的柔性光导波管的光传播损耗。光传播损耗利用光损耗测定值(db)除以光导波管长(10cm)来算出,并以表8中所示的基准进行评价。[高温高湿放置试验]针对所获得的柔性光导波管,使用高温高湿试验机(爱斯佩克(espec)(股份)制造的“pl-2kt”),在依据jpca规格(jpca-pe02-05-01s)的条件下,实施温度85℃、湿度85%的高温高湿放置试验1000小时。使用与所述相同的光源、光接收元件、入射光纤、及出射光纤测定实施高温高湿放置试验后的光导波管的光传播损耗,并以表8中所示的基准进行评价。[温度循环试验]针对所获得的柔性光导波管,使用温度循环试验机(楠本化成(股份)制造的“爱泰克闻亭(etacwintech)nt1010”),在依据jpca规格(jpca-pe02-05-01s)的条件下,实施温度-55℃与125℃之间的温度循环试验1000次。将详细的温度循环试验条件示于表6中。[表6]使用与所述相同的光源、光接收元件、入射光纤、及出射光纤测定实施温度循环试验后的光导波管的光传播损耗,并以表8中所示的基准进行评价。[回流试验]针对所获得的柔性光导波管,使用回流试验机(古河电气工业(股份)制造的“沙罗曼达(salamander)xna-645pc”),在依据电子电路互连与封装协会(theinstituteoftheinterconnectingandpackingelectroniccircuit,ipc)/jedecj-std-020b的条件下,在氮气环境下实施最高温度为265℃的回流试验3次。将详细的回流条件示于表7中,将回流炉内的温度分布示于图2中。[表7]使用与所述相同的光源、光接收元件、入射光纤、及出射光纤测定实施回流试验后的光导波管的光传播损耗,并以表8中所示的基准进行评价。[表8]评价◎0.1db/cm以下○大于0.1db/cm、且为0.2db/cm以下δ大于0.2db/cm、且为0.3db/cm以下×大于0.3db/cm将对于实施例31中所获得的柔性光导波管的评价示于表9中。[表9]评价光传输损耗○高温高湿放置试验○温度循环试验○回流试验○[产业上的可利用性]自本发明的末端改性可溶性多官能乙烯基芳香族共聚合物或含有其的材料所获得的硬化物因耐热性、相容性及韧性得到改善,且低介电特性优异,故作为各种电气绝缘材料或面向层叠体的材料有用。另外,本发明的硬化性树脂组合物的透明性、耐热性、强韧性优异,能够形成高精度的厚膜,不仅作为透明材料中的硬化性树脂组合物有用,而且尤其在光导波管形成用途中有用。[符号的说明]1:光导波管2:芯部3:上部包层4:下部包层5:基材当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1