布瑞哌唑的制备方法及用于制备布瑞哌唑的化合物与流程
本发明属于药物合成技术领域,提供了一种布瑞哌唑的制备方法及用于制备布瑞哌唑的化合物。
背景技术:
布瑞哌唑(brexpiprazole),化学名:7-(4-(4-(苯并[b]噻吩-4-基-哌嗪-1-基)丁氧基)-1h-喹啉-2-酮,由大塚制药株式会社研发,是一多巴胺d2受体部分激动剂(改善阳性和阴性症状,认知障碍及抑郁症状),5-ht2a受体拮抗剂(改善阴性症状,认知功能障碍,抑郁症的症状,失眠),xl肾上腺素受体拮抗剂(改善精神分裂症阳性症状),5-羟色胺摄取/再摄取抑制剂(改善抑郁症状);同时,又是5-ht1a部分激动剂(抗焦虑和抗抑郁活性)和5-ht7拮抗剂作。其于20150711获美国fda批准用于精神分裂症,重度抑郁症的辅助治疗。布瑞哌唑具有以下结构:
目前有很多文献专利也公开报道了布瑞哌唑的合成方法。大塚制药株式会社在pct申请wo2006112464a1中公开了布瑞哌唑的制备路线,见附图1。该路线的难点在于第一步反应生成了不易分离的副产物,通过柱层析也不易得到高纯度的中间体,从而影响了最终产品布瑞哌唑的纯度和收率。
随后,大塚制药株式会社在pct申请wo2013015456a1中公开了这步反应的另一制备方法,见附图2,该路线需要用钯化合物和叔膦或钯卡宾络合物,这些试剂都比较昂贵,成本高,对环境不友好且不适合工业化大生产。
wo2015054976a1公开了布瑞哌唑的关键中间体1-(苯并噻吩-4-基)-哌嗪及其盐的制备,见附图3,其中r为酰基类氨基保护基如甲酰基、乙酰基等,或烷氧羰基类氨基保护基如叔丁氧羰基、苄氧羰基等。该路线以2-氯-6-氟苯甲醛为原料经过5步合成1-(苯并噻吩-4-基)-哌嗪盐酸盐,虽然其操作简便,试剂易得,但路线较为繁琐造成生产周期长,收率较低。
鉴于现有布瑞哌唑制备方法存在的局限性,因此有必要寻找一条经济、实用、环保的新路线,以提高工艺稳定性,降低成本、缩短生产周期、提高产品质量。
技术实现要素:
:
本发明的目的是提供一种布瑞哌唑的制备方法及用于制备布瑞哌唑的化合物,以克服现有技术存在的上述缺陷。
因而,根据本发明的一方面,本发明提供一种式ⅳ所示的化合物或其盐,
其中,所述盐选自盐酸盐、氢溴酸盐、磺酸盐、甲磺酸盐、对甲苯磺酸盐、草酸盐、磷酸盐或马来酸盐中的一种。
发明人惊奇的发现,式ⅳ化合物可以作为制备布瑞哌唑的关键中间体,在使用式ⅳ化合物制备布瑞哌唑时,使用式ⅳ化合物只需一步即可得到布瑞哌唑,该方法制备的布瑞哌唑收率高,且产物纯度高。另外由起始原料获得式ⅳ化合物的工艺简单,且原料易得,适宜工业化生产。
根据本发明的另一方面,本发明提供一种式ⅳ化合物的制备方法,包括如下步骤:
(1)在有溶剂中或没有溶剂的情况下,碱性物质存在下,式ⅰ化合物与式ⅶ化合物反应,以便获得式ii化合物,
(2)式ⅱ化合物与式ⅵ化合物(7-羟基喹啉酮)反应以便获得式ⅲ化合物,
(3)式ⅲ化合物脱保护基r以获得式ⅳ化合物,
其中,式ⅰ、式ii、式iii化合物中的r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种;
式ⅶ化合物中x1、x2分别为卤素,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴、氯。
式ii化合物中的x3为卤素或羟基,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴;x3优选为溴、羟基。
根据本发明的另一方面,本发明提供一种由式ⅳ制备式ⅴ化合物(布瑞哌唑)的制备方法,包括式ⅳ化合物可以进一步与式ⅷ化合物反应,以便获得式ⅴ化合物,
其中,式ⅷ中的x4为卤素;
根据本发明的另一方面,本发明提供一种由式ⅰ为起始原料,制备式ⅴ的方法,包括如下步骤:
(1)在有溶剂中或没有溶剂的情况下,碱性物质存在下,式ⅰ化合物与式ⅶ化合物反应,以便获得式ii化合物,
(2)式ⅱ化合物与式ⅵ化合物(7-羟基喹啉酮)反应以便获得式ⅲ化合物,
(3)式ⅲ化合物脱保护基r以获得式ⅳ化合物,
(4)式ⅳ化合物可以进一步与式ⅷ化合物反应,以便获得式ⅴ化合物,
其中,式ⅷ中的x4为卤素;
其中,式ⅰ、式ii、式iii化合物中的r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种。
式ⅶ化合物中x1、x2分别为卤素,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴、氯。
式ii化合物中的x3为卤素或羟基,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴;x3优选为溴、羟基。
根据本发明的另一方面,本发明提供一种式ⅱ所示化合物或其盐、式ⅲ所示化合物或其盐;
其中,r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种;
式ii化合物中的x3为卤素或羟基,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴;x3优选为羟基;
任选地,所述盐为选自盐酸盐、氢溴酸盐、磺酸盐、甲磺酸盐、对甲苯磺酸盐、草酸盐、磷酸盐和马来酸盐的至少一种。
也即,本发明提供制备布瑞哌唑(式ⅴ化合物)的化合物,式ⅱ所示化合物或其盐、式ⅲ所示化合物或其盐。由此,基于这些化合物制备式ⅳ,进而制备布瑞哌唑。
具体实施例:
根据本发明的一个方面,本发明提供一种式ⅳ所示的化合物或其盐,
其中,所述盐选自盐酸盐、氢溴酸盐、磺酸盐、甲磺酸盐、对甲苯磺酸盐、草酸盐、磷酸盐或马来酸盐中的一种。
根据本发明的一个方面,本发明提供一种式ⅳ化合物的制备方法,包括在质子极性溶剂中,经布朗特酸或无机碱催化下,式ⅲ化合物脱保护基r以获得式ⅳ化合物,
其中,式iii化合物中的r为氨基保护基;
任选地,所述氨基保护基为酰基、烷氧羰基中的一种;
任选地,所述酰基选自甲酰基、乙酰基或丙酰基中的一种;
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种。
在制备式ⅳ化合物的实施例中,所述的质子极性溶剂选自c1-c4直链或支链烷醇、短链脂肪酸或水中的一种或几种;所述的布朗特酸选自盐酸、氯化氢、氢溴酸、磷酸、三氟乙酸中的一种或几种,无机碱选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾或氢氧化钠中的一种或几种,所述反应在10℃-90℃的温度下进行,所述温度优选为20℃-60℃。
任选地,所述的c1-c4直链或支链烷醇优选自乙醇、丙醇、异丙醇、正丁醇、异丁醇中的一种或几种;任选地,所述的短链脂肪酸优选自甲酸、乙酸、丙酸、丁酸中的一种或几种。
实施例中,式ⅲ化合物是通过如下步骤制备获得的:在有机溶剂中,碱性物质存在下,式ⅱ化合物与式ⅵ化合物(7-羟基喹啉酮)反应以便获得式ⅲ化合物,
其中,式ii化合物中的r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种;
式ii化合物中的x3为卤素或羟基,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴;x3优选为溴、羟基。
在式ⅲ化合物制备的具体实施例中,有机溶剂为非质子极性溶剂或者非质子性极性溶剂与非极性溶剂的混合物,所述的碱性物质为选自碳酸钾、碳酸钠、氢氧化钠、氢氧化钾,优选碳酸钾、氢氧化钾中的一种或几种;所述的非质子极性溶剂选自二甲基甲酰胺、二甲基乙酰胺、四氢呋喃、甲基四氢呋喃、丙酮、乙腈、二甲基亚砜、六甲基磷酰胺、吡啶或甲基吡啶中的一种或几种,所述的非极性溶剂选自二甲苯、4-甲基-2-戊酮、甲苯、乙苯或异丙苯中的一种或几种;反应在20℃-150℃的温度下进行,所述的温度优选为100℃-140℃,更优选120℃-125℃。
实施例中,所述的式ii化合物是通过如下步骤制备获得的:在有溶剂中或没有溶剂的情况下,碱性物质存在下,式ⅰ化合物与式ⅶ化合物反应,以便获得式ii化合物,
其中,式ⅰ化合物中的r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种;
式ⅶ化合物中x1、x2分别为卤素,卤素为选自氟、氯、溴和碘的至少一种,优选溴、氯。
在式ii化合物制备的具体实施例中,所述的有机溶剂选自乙腈、丙酮、丁酮、乙醇、甲醇中的一种或几种混合物,或上述溶剂中的一种或几种与水的混合物;碱性物质选自碳酸钾、碳酸钠、碳酸氢钾和碳酸氢钠中个一种或几种混合物。
实施例中,式ⅳ化合物进一步制备式ⅴ化合物,制备通过如下步骤:在存在钯化合物和叔膦时,碱性物质存在下,在质子性溶剂中或没有溶剂的情况下,式ⅳ化合物可以进一步与式ⅷ化合物反应,以便获得式ⅴ化合物,
其中,式ⅷ中的x4为卤素;任选地,所述卤素为选自氟、氯、溴和碘的至少一种;
在制备式ⅴ化合物的具体实施例中,钯化合物选自醋酸钯、氯化钯,所述的叔膦选自三叔丁基四苯基硼酸磷鎓、2-二环己基膦基-2’,6’-二甲氧基-1,1’-联苯、2-(二叔丁基膦基)-1,1’-联苯、2-(二叔丁基膦基)-2’-甲基-1,1’-联苯、2-(二叔丁基膦基)1,1’-联萘、2-二环己基膦基-2’,6’-二异丙氧基-1,1’-联苯、n-苯基-2-(二叔丁基膦基)吡咯和1-苯基-2-(二叔丁基膦基)-1h-茚中的一种或几种;碱性物质可以使用碳酸钾、碳酸钠、氢氧化钠、氢氧化钾、醋酸钠或醋酸钾中的一种或几种;质子性溶剂选自c1-c4直链或支链烷醇、芳烃、醚类、酮类中的一种或几种,反应温度为50℃-140℃,优选110℃-135℃;。
在制备式ⅴ化合物的优选实施例中,质子性溶剂选自乙醇、丙醇、异丙醇、正丁醇、异丁醇、甲苯、二甲苯、乙苯、异丙苯中、自四氢呋喃、乙二醇二甲醚、1,4-二氧六环、乙二醇单甲醚、乙二醇单乙醚、甲基四氢呋喃、丁酮、4-甲基-2-戊酮、环己酮或丙酮中的一种或几种,反应温度为50℃-140℃,优选110℃-135℃;。
实施例中,式ⅱ所示化合物或其盐、式ⅲ所示化合物或其盐;
其中,r为氨基保护基,
任选地,所述氨基保护基为酰基、烷氧羰基中的一种,
任选地,所述酰基选自甲酰基、乙酰基和丙酰基中的一种,
任选地,所述烷氧羰基优选甲氧羰基、乙氧羰基、叔丁氧羰基和苄氧羰基中的一种;
式ii化合物中的x3为卤素或羟基,任选地,所述卤素为选自氟、氯、溴和碘的至少一种,优选溴;x3优选为羟基。
任选地,所述盐为选自盐酸盐、氢溴酸盐、磺酸盐、甲磺酸盐、对甲苯磺酸盐、草酸盐、磷酸盐和马来酸盐的至少一种。
一般合成路线:
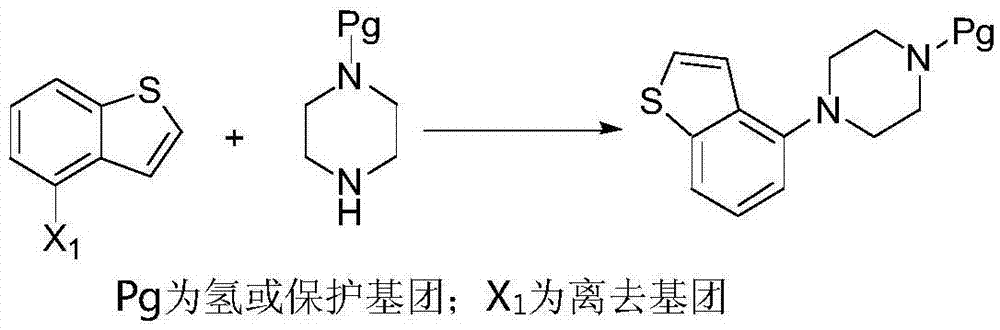
本发明公开的方法以式ⅰ化合物和1,4-二卤代丁烷为原料先合成螺环类化合物(式ⅱ化合物),式ii化合物与7-羟基喹啉酮反应生成式iii化合物,式iii化合物脱保护基后得式ⅳ化合物,最后式ⅳ化合物与4-卤代苯并噻吩反应得到目标产物布瑞哌唑(式ⅴ化合物)。
实验路线如下:
其中,上述反应路线中的r、x1、x2、x3、x4如上文所定义的。
本发明的有益效果:
本发明的制备布瑞哌唑方法,包括采用价格低廉的市售化工原料及操作简便的合成工艺制备布瑞哌唑,易于大规模经济化地生产布瑞哌唑,而且工艺的化学反应的原子利用率高,更利于绿色化学的发展。
附图说明
图1为wo2006112464a1中公开了布瑞哌唑的制备路线图。
图2为wo2013015456a1中公开的wo2006112464a1的第一步反应的另一种制备路线图。
图3:wo2015054976a1公开的布瑞哌唑的关键中间体1-(苯并噻吩-4-基)-哌嗪及其盐的制备路线。
下面将结合实施例对本发明的方案进行解释。本领域技术人员将会理解,下面的实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体技术或条件的,按照本领域内的文献所描述的技术或条件或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
具体实施方式
实施例18-boc-5,8-二氮杂螺[4,5]癸烷(式ⅱ化合物,r=-boc,x3=-oh)的制备
将n-boc哌嗪(37.2g,0.2mol)、1,4-二溴丁烷(43.2g,0.2mol)、碳酸钾(55.2g,0.4mol)、乙腈(225g)和水(16g)依次投入1l三口瓶中,升温至回流(76-78℃)。20h后反应进程通过气相色谱监测,若气相检测n-boc哌嗪含量较低且不再变化时停止反应,趁热过滤,浓缩乙腈后用异丙醇重结晶,得白色固体ⅱ32.3g,收率72.8%。所得产品通过质谱、核磁、红外进行结构确认为化合物ⅱ(r=-boc,x3=-oh)。ms:rt=0.37min,[m-h]=257.2,[m-oh]=241.1;
1hnmr(400mhz,meod),1.51(s,9h);2.26(s,4h);3.51-3.54(m,4h);3.69-3.72(t,j=6.0hz,4h),3.81(s,4h).ir:2978.66cm-1(c-h(ch3));1701.01cm-1(c=o);1145.79cm-1(c-o-c)。
实施例28-boc-5,8-二氮杂螺[4,5]癸烷(式ⅱ化合物,r=-boc,x3=-oh)的制备
将n-boc哌嗪(37.2g,0.2mol)、1,4-二氯丁烷(25.4g,0.2mol)、碳酸氢钠(35g)、乙醇(110g)和水(16g)依次投入1l三口瓶中,升温至回流。20h后反应进程通过气相色谱监测,若气相检测n-boc哌嗪含量较低且不再变化时停止反应,趁热过滤,浓缩除去溶剂后用异丙醇重结晶,得白色固体ⅱ32.3g,收率72.8%。所得产品通过质谱、核磁、红外进行结构确认为化合物ⅱ(r=-boc,x3=-oh)。ms:rt=0.37min,[m-h]=257.2,[m-oh]=241.1;1hnmr(400mhz,meod),1.51(s,9h);2.26(s,4h);3.51-3.54(m,4h);3.69-3.72(t,j=6.0hz,4h),3.81(s,4h).ir:2978.66cm-1(c-h(ch3));1701.01cm-1(c=o);1145.79cm-1(c-o-c)。
实施例38-乙氧羰基-5,8-二氮杂螺[4,5]癸烷螺环化合物(式ⅱ化合物,r=-cooch2ch3,x3=-oh)的制备
将n-乙氧羰基哌嗪(31.64g,0.2mol)、1-溴-4-氯丁烷(34.3g,0.2mol)、碳酸钾(55.2g,0.4mol)、乙腈(225g)和水(16g)依次投入1l三口瓶中,升温至回流(76-78℃)。反应进程通过气相色谱监测,若气相检测n-boc哌嗪含量较低且不再变化时(22h左右)停止反应,反应停止后趁热过滤,浓缩乙腈后用异丙醇重结晶,得白色固体26.8g(ⅱ,r=-cooch2ch3,x3=-oh),收率58.2%。
实施例47-[4-(4-boc-1-哌嗪)丁氧基]-2(1h)-喹啉酮(式ⅲ化合物,r=-boc)的制备
将实施例1所得螺环ⅱ(r=-boc,x3=-oh)(折算纯度后)(7.7g,0.024mol)、7-羟基喹啉酮(3.2g,0.02mol)、碳酸钾(5.5g,0.04mol)、4-甲基-2-戊酮(mibk75.7g)和二甲基乙酰胺(dmac,11.4g)加入250ml三口瓶中,升温至微回流,内温约116-118℃。反应20h左右薄层色谱检测,7-羟基喹啉酮剩余较少且含量不再变化时,停止反应,先蒸去溶剂,然后加水搅拌,用二氯甲烷提取,5%氢氧化钠溶液洗涤、水洗,脱去溶剂,得淡黄色固体8.9g,收率77.9%。所得产品通过质谱、核磁、红外进行结构确认为ⅲ(r=-boc)。ms:rt=1.697min,[m+h]=402.2.
1hnmr(400mhz,meod),1.47(s,9h);1.71-1.78(m,2h);1.84-1.90(m,2h);2.45-2.49(m,6h);3.45(s,4h);4.10-4.13(t,j=6.1hz,2h);6.44-6.46(d,j=9.4hz,1h);6.85-6.90(m,2h);7.55-7.58(d,j=8.7hz,1h);7.88-7.90(d,j=9.4hz,1h).
ir:3381.22cm-1(n-h);2825.28cm-1[c-h(ch3)];1652.34cm-1,1626.06cm-1(c=o);
1220.73cm-1,1137.02cm-1(c-o-c).
实施例57-[4-(4-boc-1-哌嗪)丁氧基]-2(1h)-喹啉酮(式ⅲ化合物,r=-boc)的制备
将实施例1所得螺环ⅱ(r=-boc,x3=-oh)(折算纯度后)(7.7g,0.024mol)、7-羟基喹啉酮(3.2g,0.02mol)、氢氧化钾(2.24g,0.04mol)、4-甲基-2-戊酮(75.7g)和四氢呋喃(5g)加入250ml三口瓶中,升温至微回流,内温约116-118℃。反应20h左右薄层色谱检测,7-羟基喹啉酮剩余较少且含量不再变化时,停止反应,先蒸去溶剂,然后加水搅拌,用二氯甲烷提取,5%氢氧化钠溶液洗涤、水洗,脱去溶剂,得淡黄色固体8.9g,收率77.9%。所得产品通过质谱、核磁、红外进行结构确认为ⅲ(r=-boc)。ms:rt=1.697min,[m+h]=402.2.1hnmr(400mhz,meod),1.47(s,9h);1.71-1.78(m,2h);1.84-1.90(m,2h);2.45-2.49(m,6h);3.45(s,4h);4.10-4.13(t,j=6.1hz,2h);6.44-6.46(d,j=9.4hz,1h);6.85-6.90(m,2h);7.55-7.58(d,j=8.7hz,1h);7.88-7.90(d,j=9.4hz,1h).
ir:3381.22cm-1(n-h);2825.28cm-1[c-h(ch3)];1652.34cm-1,1626.06cm-1(c=o);
1220.73cm-1,1137.02cm-1(c-o-c).
实施例67-[4-(4-boc-1-哌嗪)丁氧基]-2(1h)-喹啉酮(式ⅲ化合物,r=-boc)的制备
将n-boc哌嗪(37.2g,0.2mol)、1,4-二溴丁烷(43.2g,0.2mol)、碳酸钾(55.2g,0.4mol)、乙醇(225g)依次投入1l三口瓶中,升温至回流(76-78℃)。20h后反应进程通过气相色谱监测,若气相检测n-boc哌嗪含量较低且不再变化时停止反应,趁热过滤,浓缩乙醇后。将7-羟基喹啉酮(16g,0.1mol)、mibk(375.7g)和dmac(55.4g)加入,升温至微回流,内温约116-118℃。反应20h左右薄层色谱检测,7-羟基喹啉酮剩余较少且含量不再变化时,停止反应,先蒸去溶剂,然后加水搅拌,用二氯甲烷提取,5%氢氧化钠溶液洗涤、水洗,脱去溶剂,得淡黄色固体(ⅲ,r=-boc)41g,收率72%。
实施例77-[4-(4-乙氧羰基-1-哌嗪)丁氧基]-2(1h)-喹啉酮(ⅲ,r=-cooch2ch3)的制备将n-乙氧羰基哌嗪(4.24g,0.025mol)、1,4-二溴丁烷(5.3g,0.025mol)、碳酸钾(6.2g,0.05mol)、丙酮(25g)和水(1.6g)依次投入1l三口瓶中,升温至回流。反应进程通过气相色谱监测,若气相检测n-乙氧羰基哌嗪含量较低且不再变化时(22h左右)停止反应,反应停止后趁热过滤浓缩干,加入7-羟基喹啉酮(4.0g,0.025mol)、碳酸钾(8.5g,0.04mol)mibk(75.7g)和dmac(11.4g),升温至微回流,内温约116-118℃。反应20h左右薄层色谱检测,7-羟基喹啉酮剩余较少且含量不再变化时,停止反应,先蒸去溶剂,然后加水搅拌,用二氯甲烷提取,5%氢氧化钠溶液洗涤、水洗,脱去溶剂,得淡黄色固体(ⅲ,r=-cooch2ch3)5.1g,收率69.1%。
实施例87-[4-(4-乙氧羰基-1-哌嗪)丁氧基]-2(1h)-喹啉酮(ⅲ,r=-cooch2ch3)的制备
将实施例3所得螺环ⅱ(r=-cooch2ch3,x3=-oh)(折算纯度后)(5.5g,0.024mol)、7-羟基喹啉酮(3.2g,0.02mol)、碳酸钾(5.5g,0.04mol)mibk(75.7g)和dmac(11.4g)加入250ml三口瓶中,升温至微回流,内温约116-118℃。反应20h左右薄层色谱检测,7-羟基喹啉酮剩余较少且含量不再变化时,停止反应,先蒸去溶剂,然后加水搅拌,用二氯甲烷提取,5%氢氧化钠溶液洗涤、水洗,脱去溶剂,得淡黄色固体(ⅲ,r=-cooch2ch3)5.0g,收率67.3%。
实施例97-[4-(哌嗪-1)丁氧基]-2(1h)-喹啉酮(ⅳ)的制备
向化合物ⅲ(r=-boc)(折算纯度后)(4.0g,0.01mol)中加入40ml乙醇,10ml浓盐酸,然后混合液于20℃下进行搅拌,30mins后薄层色谱检测,若反应完全则停止反应。用50%氢氧化钠溶液中和,蒸去乙醇,然后每次用20ml二氯甲烷,萃取3次,干燥浓缩得黄色固体2.85g,经hplc检测纯度为95%,收率95%,产品通过质谱、核磁、红外进行结构确认为化合物ⅳ。
ms:rt=0.883min,[m+h]=302.2.
1hnmr(400mhz,meod),1.75-1.77(d,j=6.8hz,2h);1.87-1.88(d,j=6.8hz,2h);2.50-2.54(m,2h);2.62(s,4h);3.04(s,4h);4.11-4.14(t,2h);6.44-6.46(d,j=9.4hz,1h);6.88-6.90(d,j=9.0hz,2h);7.58-7.60(d,j=8.3hz,1h);7.90-7.92(d,j=9.4hz,1h).
ir:2946.16cm-1(n-h);1639.60cm-1(c=o);1221.29cm-11(c-o-c).
实施例107-[4-(哌嗪-1)丁氧基]-2(1h)-喹啉酮(ⅳ)的制备
室温下向化合物ⅲ(r=-cooch2ch3)(3.7g,0.01mol)中加入5.5克氢氧化钠与42ml甲醇的混合物,反应混合物在回流状态下反应12h,hplc检测化合物ⅲ(r=-cooch2ch3)含量低于0.5%后,浓缩除去甲醇,加入20毫升水,然后每次用20ml二氯甲烷,萃取3次,氯化钠溶液洗脱溶得黄色固体(ⅳ)2.52g,收率84%。产品通过质谱、核磁、红外进行结构确认为化合物ⅳ。
ms:rt=0.883min,[m+h]=302.2.
1hnmr(400mhz,meod),1.75-1.77(d,j=6.8hz,2h);1.87-1.88(d,j=6.8hz,2h);2.50-2.54(m,2h);2.62(s,4h);3.04(s,4h);4.11-4.14(t,2h);6.44-6.46(d,j=9.4hz,1h);6.88-6.90(d,j=9.0hz,2h);7.58-7.60(d,j=8.3hz,1h);7.90-7.92(d,j=9.4hz,1h).
ir:2946.16cm-1(n-h);1639.60cm-1(c=o);1221.29cm-11(c-o-c).
实施例117-[4-(哌嗪-1)丁氧基]-2(1h)-喹啉酮(ⅳ)的制备
室温下向化合物ⅲ(r=-cooch2ch3)(3.7g,0.01mol)中加入5.5克氢氧化钠与25ml80%的乙醇的混合物,反应混合物在10℃反应12h,真空浓缩除去乙醇,然后每次用20ml二氯甲烷,萃取3次,氯化钠溶液洗脱溶得黄色固体(ⅳ)2.58g,收率86%,hplc检测纯度99.1%。产品通过质谱、核磁、红外进行结构确认为化合物ⅳ。
ms:rt=0.883min,[m+h]=302.2.
1hnmr(400mhz,meod),1.75-1.77(d,j=6.8hz,2h);1.87-1.88(d,j=6.8hz,2h);2.50-2.54(m,2h);2.62(s,4h);3.04(s,4h);4.11-4.14(t,2h);6.44-6.46(d,j=9.4hz,1h);6.88-6.90(d,j=9.0hz,2h);7.58-7.60(d,j=8.3hz,1h);7.90-7.92(d,j=9.4hz,1h).
ir:2946.16cm-1(n-h);1639.60cm-1(c=o);1221.29cm-11(c-o-c).
实施例127-[4-(哌嗪-1)丁氧基]-2(1h)-喹啉酮(ⅳ)的制备
向化合物ⅲ(r=-boc)(折算纯度后)(4.0g,0.01mol)中加入30ml冰乙酸,5毫升浓盐酸,然后混合液于20℃下进行搅拌,30mins后薄层色谱检测,若反应完全则停止反应。用50%氢氧化钠溶液中和,然后每次用20ml二氯甲烷,萃取3次,干燥浓缩得黄色固体2.85g,经hplc检测纯度为95%,收率95%,产品通过质谱、核磁、红外进行结构确认为化合物ⅳ。
ms:rt=0.883min,[m+h]=302.2.
1hnmr(400mhz,meod),1.75-1.77(d,j=6.8hz,2h);1.87-1.88(d,j=6.8hz,2h);2.50-2.54(m,2h);2.62(s,4h);3.04(s,4h);4.11-4.14(t,2h);6.44-6.46(d,j=9.4hz,1h);6.88-6.90(d,j=9.0hz,2h);7.58-7.60(d,j=8.3hz,1h);7.90-7.92(d,j=9.4hz,1h).
ir:2946.16cm-1(n-h);1639.60cm-1(c=o);1221.29cm-11(c-o-c).
实施例137-[4-(4-氯苯并[b]噻吩)-4-哌嗪-1)丁氧基]-2(1h)-喹啉酮(式ⅴ化合物)盐酸盐的制备
在120-130℃下将4-氯苯并[b]噻吩(32.8mmol,5.5g)、哌嗪化合物ⅳ(30.3mmol,9.0g)、醋酸钯(0.012mmol,2.7mg),三叔丁基四苯基硼酸磷鎓(0.012mol,6.2mg)、碳酸钾(88.95mmol,12.3g)和二甲苯(70ml)搅拌5h,检测哌嗪化合物ⅳ基本消失后,将反应液冷却至室温后,向其中加水、分离,用水洗二甲苯层,然后用氯化钠溶液洗,加活性炭,在室温下搅拌30mins后过滤,滤液中加浓盐酸,在室温下对得到的混合物搅拌30mins,过滤干燥得盐酸盐,黄色固体11.6g,收率81%,hplc检测纯度99.5%。
实施例147-[4-(4-氯苯并[b]噻吩)-4-哌嗪-1)丁氧基]-2(1h)-喹啉酮的制备
将7-[4-(4-氯苯并[b]噻吩)-4-哌嗪-1)丁氧基]-2(1h)-喹啉酮(式ⅴ化合物)盐酸盐10克加至90毫升二氯甲烷中,加15克碳酸钾,45毫升水,搅拌2小时,分出二氯甲烷层,水层以20毫升二氯甲烷萃取一次,合并二氯甲烷层,用水洗两次,每次30毫升,二氯甲烷层浓缩除去二氯甲烷,得到微黄色固体8.8g,收率96%,hplc检测纯度99.6%。
实施例157-[4-(4-氯苯并[b]噻吩)-4-哌嗪-1)丁氧基]-2(1h)-喹啉酮(式ⅴ化合物)盐酸盐的制备
在65-70℃下将4-氯苯并[b]噻吩(32.8mmol,5.5g)、哌嗪化合物ⅳ(30.3mmol,9.0g)、醋酸钯(5.6mg),2-二环己基膦基-2’,6’-二异丙氧基-1,1’-联苯(69mg)、碳酸钾(88.95mmol,12.3g)和四氢呋喃(80ml)搅拌5h,检测哌嗪化合物ⅳ基本消失后,将反应液过滤浓缩后冷却至室温后,向其中加80毫升二甲苯、30克水搅拌30分钟,分离二甲苯层,用水洗二甲苯层,然后用氯化钠溶液洗,加活性炭,在室温下搅拌30mins后过滤,滤液中加浓盐酸,在室温下对得到的混合物搅拌30mins,过滤干燥得盐酸盐,黄色固体11.9g,收率83%,hplc检测纯度99.3%。
- 还没有人留言评论。精彩留言会获得点赞!