消旋吡喹胺的回收制备方法与流程
本发明属于药物合成领域,具体涉及抗虫药-左旋吡喹酮的制药中间体-消旋吡喹胺的回收制备方法。
背景技术:
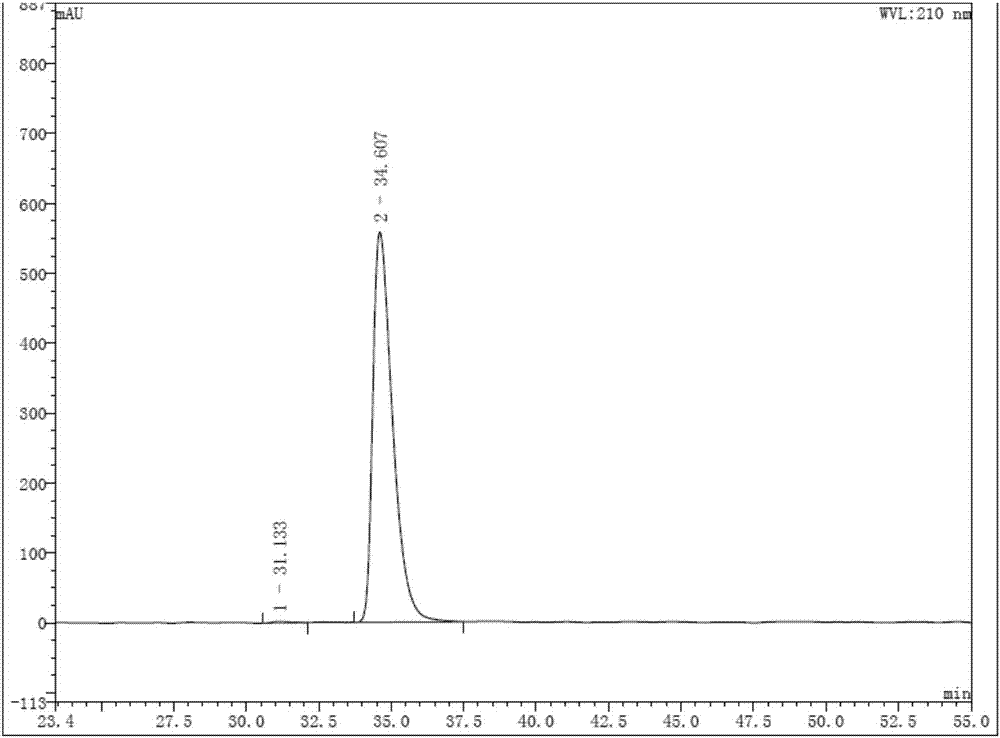
:吡喹酮(praziquantel)是一种广谱性抗寄生虫病药物,适用于日本血吸虫病、埃及血吸虫病、曼氏血吸虫病、并殖吸虫病(肺吸虫病)、华支睾吸虫病、包虫病、囊虫病、孟氏裂头蚴病、姜片虫病、缔虫病等的治疗,尤其适用于日本血吸虫病和华支睾吸虫病的治疗。1980年在德国率先以商品名称“cesol”上市后,很快成为治疗蠕虫病的首选药物。消旋体吡喹酮剂量大,不良反应明显。文献报道,左旋吡喹酮(r-3)是杀虫的有效成分,疗效优于消旋体,而右旋吡喹酮(s-3)则几乎无效,且具有一定毒副作用。左旋吡喹酮具有剂量小、疗效较高和不良反应较轻等优点。目前,科学界对左旋吡喹酮的合成进行了深入研究,主要存在的问题是成本居高不下,无法工业化生产。因此,开发左旋吡喹酮具有重要的经济和社会效益。根据文献报道,左旋吡喹酮的合成主要分为两类方法:(1)手性不对称合成其唯一的手性中心,该法使用昂贵手性催化剂或者手性辅基,总收率低,成本较高,无法工业化生产。例如,文献tetrahedron:asymmetry,17(2006),p1415-1419报道了的合成左旋吡喹酮的方法主要是使用了noyori手性催化剂来构建手性中心,具体地,采用bischler–napieralski反应构建3,4-二氢异喹啉环,然后使用noyori催化剂不对称催化还原亚胺,建立手性中心。该步关键使用到昂贵的手性催化剂,反应收率仅为52%,光学纯度仅为62%ee。且该路线总收率偏低,原料成本高,难以规模化生产,具体合成参考以下路线一。路线一文献journalofchemicalresearch;nb.3;(2004);p186-187报道的方法为:先引入手性辅基(r)-对甲苯亚磺酰基,再通过pictet–spengler反应构建四氢异喹啉环,从而诱导出手性中心。该步关键使用到相对较贵的手性辅基试剂(1s,2r,5s)-(+)-薄荷基(r)-对甲苯亚磺酸盐,且反应需在-78℃下进行,收率仅为63%。因此,该路线总收率偏低,约20%,原料成本高,反应条件苛刻,难以规模化生产。文献tetrahedron,;70(2014);p3864-3870报道的方法为:使用廉价的苯丙氨酸为原料,通过环合反应诱导出手性中心。其中,最后一步脱羧反应操作繁琐,反应条件较为苛刻,产品杂质多,需柱层析纯化,总收率仅为20%。综上看来,通过手性不对称合成来构建左旋吡喹酮的关键手性中心,要么是在合成方法中使用昂贵手性催化剂或者手性辅基,要么是手性诱导不对称合成构建,路线冗长,反应条件严苛,中间体纯化麻烦,总收率低,总生产成本较高,无法实现工业化大量生产。(2)手性拆分,即对关键中间体(如吡喹胺1)用酒石酸酯衍生物l-(-)-dbta进行拆分,获得制备左旋吡喹酮所需要的手性中心的r-1。再就r-1的仲胺氨基进行环己基甲酰化得到左旋吡喹酮,见以下路线二。路线二对消旋吡喹胺1的合成,在先前的文献中已有很多报道,这些文献多涉及了消旋吡喹酮的全合成工艺。例如:德国专利de2504250,de2508947报道的路线:以异喹啉为起始原料,经reissert反应、催化氢化、酰化、环合、水解五步反应制得消旋吡喹胺;见以下路线三。该方法工艺成熟、原料易得、成本较低。该方法曾是国内外广泛采用的吡喹酮工业生产方法。路线三另外,文献journalofheterocyclcchemistryvol23(1986)p189-190则报道了:以廉价的n-苄基亚氨基二乙酸和β-苯乙胺为原料,经环合、还原、再环合、氢化脱苄四步反应制得消旋吡喹胺;见以下路线四。此方法原料易得,反应条件温和,步骤不长,环境友好,易于规模化生产。路线四文献tetrahedronvol.54nb.26(1998)7395报道的路线:以β-苯乙胺、氯乙酰氯、氨基乙醛缩二甲醇等为原料,经酰化、取代、环合三步反应制得消旋吡喹胺;见以下路线五。该方法原料价廉易得,工艺简单,环境友好,合成步骤少,总收率高,成本低,易于工业化生产。路线五就上述拆分法制备左旋吡喹酮,会损失消旋体中的50%的不需要的构型吡喹胺s-1的关键中间体,从而导致成本大幅上升。因此,若能够将丢弃的s构型的吡喹胺重新废物利用,将其消旋化,并将得到消旋的吡喹胺用于拆分法合成左旋吡喹酮;从而提高目标产物的收率,最大限度的降低了左旋吡喹酮的生产成本。技术实现要素:本发明旨在提供一种将丢弃的s构型的吡喹胺重新废物利用的消旋化方法。本发明人课题组为此进行了多方面深入的研究:首先,对吡喹胺s‐1采用有机化学理论上常规的消旋化方法,譬如酸、碱或者加热的作用下消旋,但是我们尝试数次之后都无法获得消旋化的产物,而剧烈的条件导致了吡喹胺s‐1分解破坏生成其他杂质。无数次的实验证明,常规消旋化方法无法应用于s构型吡喹胺的消旋化工艺。经过深入的研究,本发明人大胆地创新,并成功开发出消旋吡喹胺的回收制备方法。本发明使用拆分吡喹胺-1的方法来制备左旋吡喹酮所丢弃的s-吡喹胺-1作为起始原料,在催化剂或者脱氢试剂作用下制得化合物2,进一步通过加氢还原制备得到消旋吡喹胺-1。而消旋体的吡喹胺-1用于拆分法合成左旋吡喹酮,从而建立一种有效的循环拆分合成手性吡喹酮,使不需要的构型吡喹胺s-1得到充分回收利用,能够最大限度的降低了左旋吡喹酮的生产成本。因此,更具体地,一方面,本发明提供了一种回收法制备消旋体的吡喹胺中间体-1的方法,包括以下步骤:1)将废弃物料s-吡喹胺-1作为起始原料,在催化剂或者脱氢试剂作用下制得化合物2,2)通过加氢还原化合物2制备得到消旋吡喹胺-1。化合物1为消旋体的吡喹胺,而s-1则为拆分合成左旋吡喹酮丢弃的非所需构型中间体。化合物2为6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮。在本发明的回收法制备消旋吡喹胺-1的方法中,所述步骤1)的操作为:将s-吡喹胺-1溶于第一溶剂中,并加入第一催化剂或者脱氢试剂,在适当温度下反应,即得化合物2。其中,所述第一溶剂选自上述醚类溶剂选自1,2-二甲氧基乙烷、甲基叔丁基醚、2-甲基四氢呋喃、环戊基甲醚;芳香烃类溶剂选自苯、甲苯、乙苯、二甲苯、三甲苯、对甲基异丙基苯、硝基苯、氯苯、二氯苯;烃类或者卤代烃类溶剂选自正庚烷、正辛烷、十氢萘、二氯乙烷;酯类溶剂选自乙酸乙酯、乙酸异丙酯、乙酸异丁酯;酮类溶剂选自2-丁酮、甲基异基酮;醇类溶剂选自甲醇、乙醇、异丙醇、叔丁醇、正丁醇、异丁醇;其他溶剂选自二甲亚砜、n,n-二甲基甲酰胺。所述第一溶剂进一步优选为环戊基甲醚、甲苯、二甲苯、异丙醇、正丁醇。其中,所述第一催化剂选自raney-ni、ni/c、cu/c、ru/c、pt/c、pd/c、ir、fe、zn等常见金属催化剂,脱氢试剂选自2,3-二氯-5,6-二氰基-1,4-苯醌(ddq)、氯醌、单质硫、二氧化硒或者有机硒、二氧化锰、五价氧钒化合物等有机化学中常用脱氢试剂。优选pt/c、raney-ni、pd/c。其中,所述步骤1)的反应温度60-140℃,优选80-140℃。所述步骤1)的更进一步优选条件:采用甲苯或者正丁醇作为第一溶剂,选用pd/c作为催化剂,反应温度为100~120℃。所述步骤2)的操作为:将化合物2溶于第二溶剂中,然后加入第二催化剂及加氢试剂在适当温度下反应,即制得消旋吡喹胺-1。其中,所述第二溶剂选自上述醚类溶剂选自1,2-二甲氧基乙烷、甲基叔丁基醚、2-甲基四氢呋喃、环戊基甲醚;芳香烃类溶剂选自苯、甲苯、乙苯、二甲苯、三甲苯、对甲基异丙基苯、硝基苯、氯苯、二氯苯;烃类或者卤代烃类溶剂选自正庚烷、正辛烷、十氢萘、二氯乙烷;酯类溶剂选自乙酸乙酯、乙酸异丙酯、乙酸异丁酯;酮类溶剂选自2-丁酮、甲基异基酮;醇类溶剂选自甲醇、乙醇、异丙醇、叔丁醇、正丁醇、异丁醇;其他溶剂选自二甲亚砜、n,n-二甲基甲酰胺。所述第二溶剂进一步优选为环戊基甲醚、甲苯、二甲苯、对甲基异丙基苯、异丙醇、正丁醇。其中,所述第二催化剂选自raney-ni、ni/c、cu/c、ru/c、pt/c、pd/c。优选pt/c、raney-ni、pd/c;所述加氢试剂选自氢气,甲酸,甲酸铵,三乙胺甲酸盐,水合肼,环己烯,环己二烯,环己醇,四氢化萘,四氢吡咯。第二催化剂优选pd/c,所述加氢试剂优选氢气。优选地,所述步骤2)的反应温度0-140℃,优选0-60℃。更优选地,所述步骤2)中所述加氢试剂优选氢气,氢气压力0.5~10atm,优选0.8~5atm所述步骤2)更进一步优选条件:甲苯或者正丁醇,pd/c,反应温度10~40℃,氢气。此外,步骤2)中使用的第二溶剂可以与步骤1)中使用的第一溶剂不同,也可以相同。此外,步骤2)中使用的第二催化剂可以与步骤1)中使用的第一催化剂不同,也可以相同。当步骤2)中使用的第二溶剂可以与步骤1)中使用的第一溶剂相同,且步骤2)中使用的第二催化剂可以与步骤1)中使用的第一催化剂相同时,步骤1)和步骤2)优选“一锅法”操作,即步骤1)完成后,反应液直接进行步骤2)的反应。“一锅法”是指化学合成反应中,几个步骤反应放在同一个反应容器中操作,各步骤得到的中间产物不进行分离纯化操作,进而简化反应操作工艺,节省生产成本。所述步骤2)的操作还可以为:将化合物2溶于第三溶剂中,然后加入还原剂在适当温度下反应,即制得消旋吡喹胺-1。其中,醚类溶剂选自四氢呋喃、1,2-二甲氧基乙烷、甲基叔丁基醚、2-甲基四氢呋喃、环戊基甲醚;芳香烃类溶剂选自苯、甲苯、乙苯、二甲苯、三甲苯、对甲基异丙基苯、硝基苯、氯苯、二氯苯;醇类溶剂选自甲醇、乙醇、异丙醇、叔丁醇、正丁醇、异丁醇。有机溶剂进一步优选为四氢呋喃、甲苯、二甲苯、甲醇、乙醇、异丙醇。所述还原剂选自还原剂选自bh3,nabh4,kbh4,nabh(oac)3,nabh4/lewisacid,优选bh3,nabh(oac)3;所述反应温度0-60℃,优选10-30℃。以上优选的实施方式可以自由组合,从而得到本发明各种优选的实施方式。本发明优势在于,通过不需要的构型吡喹胺s-1充分回收利用得到的消旋体化合物1,在再被利用通过拆分法制备左旋吡喹酮。从而能够变废为宝,极大地节省左旋吡喹酮的生成成本。具体实施方式通过以下实施例进一步说明本发明,以下实施例仅用于更具体的说明本发明优选的实施方案,不用于对本发明的技术方案进行限定。上述本发明的方案均为可实现本发明目的之技术方案。以下实施例所采用温度和试剂,均可以用上述相应温度和试剂替代以实现本发明之目的。在下述制备实施例中,核磁共振由varianinova型核磁共振仪测定,tms为内标,化学位移单位为ppm。对于消旋的吡喹胺1,s构型吡喹胺s-1,r构型吡喹胺r-1的构型含量检测通过以下手性hplc方法。采用手性hplc分析方法:色谱柱:250mm×4.6mm,ic‐3,3μm;紫外检测器波长:210nm;流速:0.4ml/min;柱温:30℃;流动相:正己烷(含0.1%的二乙胺):异丙醇=30:70。购买获得的消旋吡喹胺‐1(对照品)的hplc图谱如图1所示,其中横坐标为时间(单位min),纵坐标为mau峰高度:从图1中得出,s‐1保留时间34.51min,含量50.65%;r‐1保留时间30.31min,含量49.35%。本发明实施例采用的通过拆分法制备左旋吡喹酮后,废弃的s构型的吡喹胺中间体s‐1;其是本发明起始原料。s构型的吡喹胺s‐1原料的hplc图谱如图2所示:,其中横坐标为时间(单位min),纵坐标为mau峰高度:从图2中得出,s‐1保留时间34.61min,含量99.66%;r‐1保留时间31.13min,含量0.34%。按照本发明消旋化方法,将s构型的吡喹胺s‐1转化成消旋吡喹胺1产物的典型hplc图谱如图3所示:其中横坐标为时间(单位min),纵坐标为mau峰高度:其中:s‐1保留时间34.57min,含量51.13%;r‐1保留时间30.30min,含量48.87%。图3是由实施例31制备得到消旋吡喹胺1产物,所测试的手性hplc图谱,以确认产物的消旋化程度。实施例13‐32以及33制备得到消旋吡喹胺1产物,经上述手性hplc图谱测试,表明,产物的消旋化程度与实施例31制备得到的产物的消旋化程度也一致;因此,本文中不一一列出手性hplc测试图谱。实施例1-12、合成化合物2实施例1250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,100ml甲苯,氮气保护下回流反应45h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体2.0g,收率40.8%。1hnmr(cdcl3)δ:3.03(t,2h),4.23(t,2h),7.26~7.80(m,4h),7.86(s,1h),8.12(s,1h);ms(esi)m/z:199.1([m+1]+),221.1([m+na]+),237.1([m+k]+)实施例2250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,100ml二甲苯,氮气保护下140℃反应40h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体1.6g,收率32.6%。实施例3250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,100ml对甲基异丙基苯,氮气保护下150℃反应36h,冷却,过滤,将反应物经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体1.5g,收率30.6%。实施例4250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,100ml十氢萘,氮气保护下180℃反应12h,冷却,过滤,将反应物经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体1.1g,收率22.4%。实施例5250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,50ml异丙醇,氮气保护下回流反应60h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体0.89g,收率18.2%。实施例6250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g钯碳,50ml正丁醇,氮气保护下回流反应48h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体1.4g,收率28.6%。实施例7250ml反应瓶中依次加入2.5gs-吡喹胺(化合物s-1),0.25g钯碳,150ml环戊基甲醚,氮气保护下回流反应48h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体0.29g,收率11.8%。实施例8250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),0.25g铂碳,100ml甲苯,氮气保护下回流反应45h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体2.3g,收率46.9%。实施例9250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),2.0graney-ni,100ml甲苯,氮气保护下回流反应45h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体0.78g,收率15.9%。实施例10250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),11.8gddq,100ml甲苯,氮气保护下回流反应24h,冷却,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体3.8g,收率77.5%。实施例11100ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),3.0g单质硫,氮气保护下加热熔融反应8h,冷却,用50ml二氯甲烷提取反应物,过滤,减压蒸除溶剂,将所得固体经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体1.3g,收率26.5%。实施例12250ml反应瓶中依次加入5.0gs-吡喹胺(化合物s-1),6.0g二氧化硒,100ml甲苯,氮气保护下90℃反应12h,冷却,将所得反应物经柱层析(洗脱剂:乙酸乙酯/石油醚)分离,浓缩洗脱剂得目标产物,为浅绿色固体2.7g,收率55.1%。实施例13-25以及实施例26-33中制备得到的消旋化吡喹酮产物-1,测试了消旋体的hplc纯度。测试方法如下:消旋体纯度由一般的非手性柱测试,分析方法如下:色谱柱:150mm×4.6mm,xbridgec18,3.5μm;紫外检测器波长:210nm;流速:1.0ml/min;柱温:35℃;流动相为乙腈/0.1%三氟乙酸水溶液的混合溶剂采用梯度洗脱方式,详细见下表:时间(min)乙腈(%)0.1%三氟乙酸水溶液(%)059535958208020703024703024.159530595注:0.1%三氟乙酸水溶液的配置:向1l水加入1ml三氟乙酸。实施例13-25、合成化合物1实施例13在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,100ml甲苯,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度98%。1hnmr(cdcl3)δ:1.88(s,1h),2.70~3.07(m,4h),3.64(m,j=17.3hz,2h),3.75(dd,1h),4.75~4.93(m,2h),7.11~7.30(m,4h);ms(esi)m/z:203.1([m+1]+),241.1([m+k]+)实施例14在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,100ml甲苯,1atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度96%。实施例15在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,100ml甲苯,3atmh2下室温反应5h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度99%。实施例16250ml反应瓶中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,9.5g甲酸铵,100ml甲苯,回流反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.3g,收率85%,hplc纯度90%。实施例17250ml反应瓶中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,3.0g水合肼(含量80%),100ml甲苯,回流反应6h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.0g,收率78%,hplc纯度85%。实施例18250ml反应瓶中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,6.2g环己烯,100ml甲苯,回流反应3h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.7g,收率92%,hplc纯度83%。实施例19在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g铂碳,100ml甲苯,5atmh2下50℃加热反应3h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度98%。实施例20在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),2.0graney-ni,100ml甲苯,5atmh2下50℃加热反应6h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度95%。实施例21在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,100ml二甲苯,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度96%。实施例22在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,50ml异丙醇,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度98%。实施例23在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,50ml正丁醇,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度97%。实施例24在氢化反应釜中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),0.25g钯碳,50ml环戊基甲醚,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体5.0g,收率98%,hplc纯度94%。实施例25250ml反应瓶中依次加入5.0g6,7-二氢-4h-吡嗪并[2,1-a]异喹啉-4-酮(化合物2),13.4g三乙酰氧基硼氢化钠,100ml二氯甲烷,室温反应12h,反应液经naoh水溶液洗涤,无水硫酸镁干燥,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.6g,收率90%,hplc纯度87%。实施例26-33、一锅法操作工艺实施例26250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g钯碳,100ml甲苯,氮气保护下回流反应45h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.95g,收率99%,hplc纯度95%。实施例27250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g铂碳,100ml甲苯,氮气保护下回流反应45h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应2h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.97g,收率99%,hplc纯度96%。实施例28250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),2.0graney-ni,100ml甲苯,氮气保护下回流反应45h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应2h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.90g,收率98%,hplc纯度93%。实施例29250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g钯碳,100ml甲苯,氮气保护下回流反应45h,冷却,然后将氮气置换成氢气,1atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.96g,收率99%,hplc纯度97%。实施例30250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g钯碳,100ml甲苯,氮气保护下回流反应45h,冷却,然后将氮气置换成氢气,1atmh2下室温反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.98g,收率99%,hplc纯度96%。实施例31250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g钯碳,100ml正丁醇,氮气保护下回流反应24h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.99g,收率99%,hplc纯度90%。实施例32250ml反应瓶中依次加入5.0gs-吡喹胺(化合物1),0.25g钯碳,100ml异丙醇,氮气保护下回流反应60h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体4.97g,收率99%,hplc纯度96%。实施例33250ml反应瓶中依次加入2.5gs-吡喹胺(化合物1),0.25g钯碳,150ml环戊基甲醚,氮气保护下回流反应48h,冷却,然后将氮气置换成氢气,5atmh2下50℃加热反应4h,冷却,过滤,减压蒸除溶剂得目标产物,为浅黄色固体2.45g,收率98%,hplc纯度95%。对比实施例34-38采用有机化学理论上常规的消旋化方法,例如酸、碱或者加热的作用下消旋,尝试对吡喹胺s‐1进行消旋化处理;但是我们尝试数次之后都无法获得消旋化的产物,而剧烈的条件 导致了吡喹胺s‐1分解破坏生成其他杂质。无数次的实验证明,常规消旋化方法无法应用于s构型吡喹胺的消旋化工艺。实施例34100ml反应瓶中依次加入2.0gs-吡喹胺(化合物1,99%ee),0.83gkoh,30mldmso,氮气保护下60℃加热反应24h,冷却,取样经手性hplc分析显示s-吡喹胺的光学纯度仍为99%ee,因此,该方法没有消旋化效果。实施例35100ml反应瓶中依次加入2.0gs-吡喹胺(化合物1,99%ee),1.0gnaoch3,30ml甲醇,氮气保护下60℃加热反应24h,冷却,取样经手性hplc分析显示s-吡喹胺的光学纯度仍为99%ee,因此,该方法没有消旋化效果。实施例36100ml反应瓶中依次加入2.0gs-吡喹胺(化合物1,99%ee),1.33g叔丁醇钾,30ml叔丁醇,氮气保护下60℃加热反应12h,冷却,大部分原料s-吡喹胺降解生成其他杂质。实施例37100ml反应瓶中依次加入2.0gs-吡喹胺(化合物1,99%ee),2.5ml浓hcl,50ml水,70℃加热反应24h,冷却,取样经手性hplc分析显示s-吡喹胺的光学纯度仍为99%ee,且有部分原料降解生成杂质,因此,该方法没有消旋化效果。实施例38100ml反应瓶中依次加入2.0gs-吡喹胺(化合物1,99%ee),2.2g浓h2so4,50ml水,70℃加热反应24h,冷却,取样经手性hplc分析显示s-吡喹胺的光学纯度仍为99%ee,且有部分原料降解生成杂质,因此,该方法没有消旋化效果。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1