一种结晶形态的Lesinurad中间体及其制备方法与流程
本发明属于医药技术领域,具体涉及一种结晶形态的lesinurad中间体及其制备方法。
背景技术:
痛风是一种单钠尿酸盐沉积所致的晶体相关性关节病,是一种世界流行的代谢病。我国国痛风的患病率约为0.15%~0.67%。随着经济的迅速发展,人们对高蛋白食物摄入的不断增加,痛风及高尿酸血症的患病率也在不断升高。
lesinurad(结构式如式i所示),化学名称为2-[[5-溴-4-(4-环丙基-1-萘-1-基)-4h-1,2,4-三唑-3-基]硫代]乙酸,是阿斯利康公司(astrazeneca)开发的一种尿酸转运蛋白1(urat1)抑制剂,于2015年在美国批准上市,商品名zurampic,用于联合黄嘌呤氧化酶抑制剂(xols)治疗痛风相关的高尿酸血症。
lesinurad通常由式ⅱ所示的中间体水解得到,该中间体的质量将直接影响终产品lesinurad的质量。
pct专利wo2006026356公开了一种lesinurad中间体ii的制备方法,合成路线如流程1所示:
流程1
中间体iii的氨基经亚硝酸钠氧化成重氮,然后与溴仿进行溴代反应,得到的中间体ii反应液,直接经柱层析纯化,得到中间体ii,为泡沫状粘稠固体,纯度约95%。使用柱层析纯化中间体ii,不利于工业化大生产,且产品纯度低、状态差。pct专利wo2009070740和wo2011085009公开了类似方法。
pct专利wo2014008295公开了一种lesinurad中间体ii的制备方法,合成路线如流程2所示:
流程2
中间体iii的氨基经亚硝酸钾氧化成重氮,然后与溴化铜进行溴代反应。反应完毕用甲苯萃取,然后用氨水和柠檬酸钠溶液多次洗涤甲苯相,以除去铜离子,得到中间体ii粗品,纯度约85%。中间体ii不经分离和纯化,继续进行氢氧化钠水解反应制备lesinurad,由于杂质较多,给终产品lesinurad的纯化带来较大困难。
pct专利wo2014008295还公开了另外一种lesinurad中间体ii的制备方法,合成路线如流程3所示:
流程3
中间体iv和n-溴代丁二酰亚胺(nbs)在四氢呋喃(thf)中,在27-32℃进行溴代反应。反应完毕,经后处理得到的中间体ii,纯度约91%。中间体ii不经分离和纯化,继续经氢氧化钠水解反应制备lesinurad,由于杂质较多,给产品lesinurad的纯化带来较大困 难。
pct专利wo2014198241公开了一种lesinurad中间体ii的制备方法,合成路线如流程4所示:
流程4
中间体iv和1,3-二溴-5,5-二甲基海因在乙酸乙酯中回流反应,经后处理得到的中间体ii,为黄色泡沫状粘稠固体,纯度约87%,杂质较多,如不纯化,不利于后续反应中产品lesinurad的纯化。
通过溴化反应来制备中间体ii,在中间体ii中有不同位置溴代或者多溴取代的杂质残留,这些溴代杂质与中间体ii有相似的性质,不易除去,中间体ii的纯度一般低于92%。如果中间体ii不经纯化,进一步加碱进行去酯化反应生成lesinurad,不同位置溴代或者多溴取代的中间体ii杂质也进行去酯化反应,生成溴代lesinurad杂质,由于溴代杂质性状与产品lesinurad比较类似,也不易分离除去,给终产品lesinurad的纯化带来较大困难。例如wo2014008295实施例2a中,lesinurad的制备,是将制备得到的中间体ii经过后处理,经去酯化生成lesinurad钠盐,再用hbr酸调酸化,然后用乙酸乙酯多次萃取,然后再浓缩乙酸乙酯到一定体积,加正庚烷析晶得到的,步骤多,操作繁琐。
综上,现有技术未得到高纯度的、结晶形式的lesinurad中间体ii,也没有公开适合工业化生产的lesinurad中间体ii的纯化方法。由于中间体ii的纯度直接影响终产品lesinurad的纯度,因此有必要对lesinurad中间体ii的纯化方法及固态形式进行研究。
技术实现要素:
出人意料的,本发明人在对lesinurad的研究中得到了高纯度的、易于储存的结晶形态的lesinurad中间体ii,且所采用方法简单可行、利于产业化。
因此,本发明一方面提供了结晶形态的lesinurad中间体ⅱ,
优选的,所述的结晶形态的lesinurad中间体ⅱ,其差示扫描量热测定在98~102℃有峰;进一步优选的,其差示扫描量热测定在99~101℃有峰;更进一步优选的,所述的结晶形态的lesinurad中间体,其具有基本上如附图4-6任一所示的差示扫描量热图谱。所述结晶的差示扫描量热有实验误差,在一台仪器和另一台仪器之间以及一个样品和另一个样品之间,吸热峰的位置和峰值可能会略有差别,实验误差或差别的数值可能小于等于5℃,或小于等于4℃,或小于等于3℃,或小于等于2℃,或小于等于1℃,因此所述差示扫描量热吸热峰的峰位置或峰值的数值不能视为绝对的。
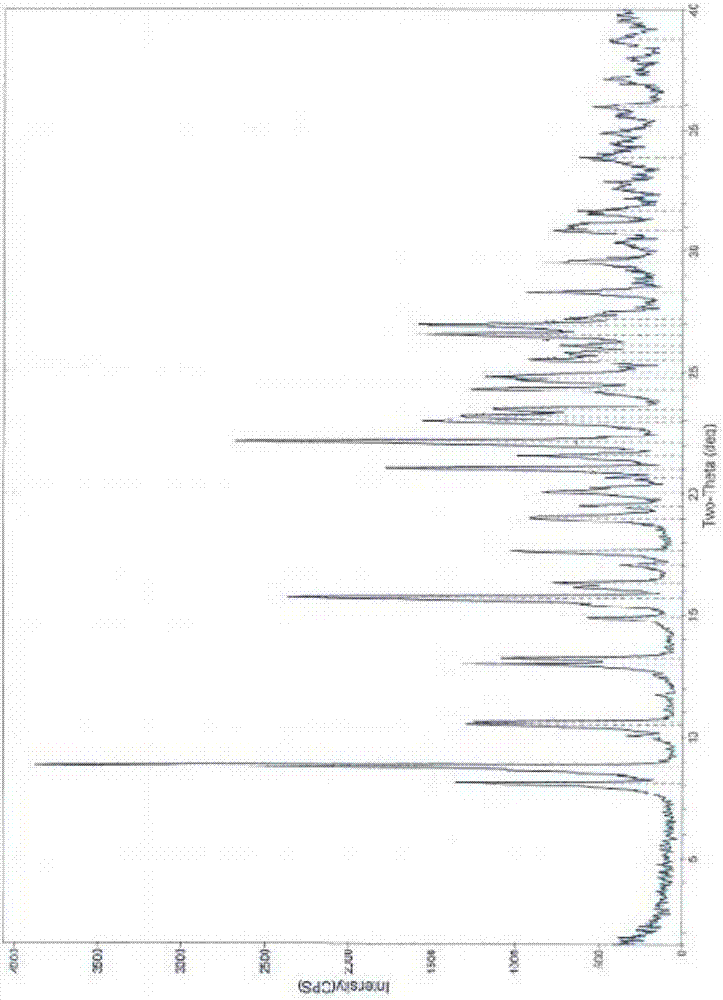
优选的,所述的结晶形态的lesinurad中间体ⅱ,使用cu-kα辐射,以2θ角度(°)表示的粉末x-射线衍射在以下位置有特征峰:8.8±0.2°、10.5±0.2°、10.6±0.2°、15.6±0.2°、22.1±0.2°、22.9±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于50%。
进一步优选的,所述的结晶形态的lesinurad中间体ⅱ,其特征在于,使用cu-kα辐射,以2θ角度(°)表示的粉末x-射线衍射在以下位置有特征峰:
(1)8.8±0.2°、10.5±0.2°、10.6±0.2°、15.6±0.2°、22.1±0.2°、22.9±0.2°、23.1±0.2°、24.8±0.2°、26.9±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于40%;或
(2)8.0±0.2°、8.8±0.2°、10.5±0.2°、10.6±0.2°、13.0±0.2°、15.6±0.2°、19.0±0.2°、22.1±0.2°、22.9±0.2°、23.1±0.2°、23.4±0.2°、24.3±0.2°、24.8±0.2°、26.5±0.2°、26.9±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于30%;或
(3)8.0±0.2°、8.8±0.2°、10.5±0.2°、10.6±0.2°、13.0±0.2°、13.2±0.2°、15.6±0.2°、19.0±0.2°、20.0±0.2°、21.0±0.2°、22.1±0.2°、22.9±0.2°、23.1±0.2°、23.4±0.2°、24.3±0.2°、24.8±0.2°、26.5±0.2°、26.9±0.2°、30.8±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于20%;或
(4)8.0±0.2°、8.8±0.2°、10.5±0.2°、10.6±0.2°、13.0±0.2°、13.2±0.2°、15.6±0.2°、17.6±0.2°、19.0±0.2°、20.0±0.2°、21.0±0.2°、21.5±0.2°、22.1±0.2°、22.9±0.2°、23.1±0.2°、23.4±0.2°、24.3±0.2°、24.8±0.2°、25.5±0.2°、26.5±0.2°、26.9±0.2°、27.2±0.2°、28.3±0.2°、29.5±0.2°、30.8±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于10%;或
(5)8.0±0.2°、8.8±0.2°、10.5±0.2°、10.6±0.2°、13.0±0.2°、13.2±0.2°、14.9±0.2°、15.6±0.2°、16.3±0.2°、17.6±0.2°、19.0±0.2°、19.5±0.2°、20.0±0.2°、21.0±0.2°、21.5±0.2°、22.1±0.2°、22.9±0.2°、23.1±0.2°、23.4±0.2°、24.3±0.2°、24.8±0.2°、25.5±0.2°、26.5±0.2°、26.9±0.2°、27.2±0.2°、28.3±0.2°、29.5±0.2°、30.8±0.2°;所述特征峰的相对峰强度(峰面积比a%)不低于5%。
进一步优选的,所述的结晶形态的lesinurad中间体ⅱ,使用cu-kα辐射,其具有基本上如附图1至3任一所示的粉末x-射线衍射图谱。术语“基本上如附图所示”是指基本上纯净的某种晶型其粉末x-射线衍射图谱中至少50%,或至少60%,或至少70%,或至少80%,或至少90%,或至少95%,或至少96%,或至少97%,或至少98%,或至少99%的峰出现在所给出粉末x-射线衍射图谱中。当样品中某种晶型的含量逐渐降低时,其粉末x-射线衍射图谱中的一些归属于该晶型的衍射峰可能会由于仪器的检测灵敏度的因素而变少。
本发明另一方面还提供一种结晶形态的lesinurad中间体ⅱ的制备方法,包括如下步骤:将lesinurad中间体ⅱ粗品溶解于溶剂中,再加入酸,然后降温至-10~30℃搅拌析晶0.5~10小时,分离,得所述的结晶形态的lesinurad中间体ⅱ。
其中:
所述溶剂选自丙酮、乙酸乙酯、丁酮、四氢呋喃、乙腈、二甲亚砜、n,n-二甲基甲酰胺、甲醇、乙醇、异丙醇、正己烷、正庚烷、石油醚中的一种或多种,优选为n,n-二甲基甲酰胺、丙酮、甲醇、乙醇、异丙醇的一种或者多种。所述溶剂可以一次加入,也可以多次加入。所述溶剂与lesinurad中间体ⅱ粗品的重量比为1~100:1,更优选为5~50:1,最优选为5~15:1。溶解温度优选为20~80℃,更优选为20~40℃。
所述酸选自硫酸、盐酸、磷酸、硝酸、氢溴酸、氢碘酸、甲酸、乙酸、丙酸、甲磺酸、乙磺酸、对甲苯磺酸、马来酸、富马酸、柠檬酸、草酸、琥珀酸和枸杞酸中的一种或多种,优选为硫酸、盐酸、磷酸和乙酸中的一种或多种。所述酸与lesinurad中间体ⅱ粗品的摩尔比为0.5~50:1,从收率考虑,更优选为0.5~5:1,最优选为1~3:1。
析晶温度优选为-5~20℃,更优选为-5~10℃,最优选为0-5℃。
所述分离步骤包括采用过滤、离心等适宜的方法将所得lesinurad中间体ⅱ结晶从结晶液中分离出来。
优选地,从去除产品中游离溶剂考虑,在分离步骤后,还包括干燥步骤,干燥方法可采用任何适宜的已知方法,优选为减压(真空)干燥。具体的干燥条件是,例如,温度优选40~60℃,更优选为40~50℃;压力优选为真空度>0.090mpa;干燥时间优选为5~20h, 更优选为5~8h。无论采用何种干燥手段,都以所得产品中溶剂残留量符合质量标准为宜。
本发明中所述lesinurad中间体ⅱ粗品,可采用现有技术中公开的任何已知方法进行制备,如下述文献公开的lesinurad中间体ⅱ的制备方法:wo2006026356、wo2009070740、wo2011085009、wo2014008295、以及wo2014198241等。
所述lesinurad中间体ⅱ粗品可为半固体或无定形固体;可以为各种纯度,优选纯度在85%以上。
本发明另一方面还提供所述的结晶形态的lesinurad中间体ⅱ在制备尿酸转运蛋白1抑制剂lesinurad中的应用。由本发明的结晶形态的lesinurad中间体ⅱ制备lesinurad的方法可参照现有技术中任何已知方法进行制备,如:wo200602635,wo2014008295。
本发明的结晶形态的lesinurad中间体ⅱ的有益效果在于:
①首次得到lesinurad中间体ⅱ结晶型固体,纯度高,大于98%;
②操作简单,不需柱层析纯化,适于工业化;
③性质稳定,便于贮存运输及后续投料操作,由其制备所得终产品lesinurad的纯度高,大于99%。
附图说明
图1:本发明实施例1所得结晶形态的lesinurad中间体ⅱ的粉末x-射线衍射图。
图2:本发明实施例2所得结晶形态的lesinurad中间体ⅱ的粉末x-射线衍射图。
图3:本发明实施例3所得结晶形态的lesinurad中间体ⅱ的粉末x-射线衍射图。
图4:本发明实施例1所得结晶形态的lesinurad中间体ⅱ的差示扫描量热图谱。
图5:本发明实施例2所得结晶形态的lesinurad中间体ⅱ的差示扫描量热图谱。
图6:本发明实施例3所得结晶形态的lesinurad中间体ⅱ的差示扫描量热图谱。
具体实施方式
以下实施例中所得样品粉末x-射线衍射图谱和差示扫描量热图谱检测条件如下:
1、粉末x-射线衍射图谱
设备名称:d/max-2500x-射线衍射仪
靶:cu(40kv,150ma)
阶跃角:0.02°
扫描范围:1.5~40.0°
2、差示扫描量热图谱
设备名称:perkinelmerdiamond差示扫描量热仪(dsc)
升温速率:20.00℃/min
制备例:lesinurad中间体ii粗品的制备(参照wo2014198241)
在2.5l三口瓶中,加40glesinurad中间体iv(0.118mol)、33.7g1,3-二溴-5,5-二甲基海因(0.118mol)和1l乙酸乙酯,回流搅拌直至反应完全。反应液冷却至室温,加1l水洗涤,分相。有机相用1l2%的亚硫酸氢钠溶液洗涤,然后分别用1l6.5%的碳酸氢钠溶液和1l水洗涤。有机相减压浓缩,得到44.6g泡沫状黄色固体,收率90.3%,纯度87.1%,xrd检测为无定形固体。
实施例1:lesinurad中间体ii结晶的制备
在100ml三口瓶中,加10glesinurad中间体ii粗品(23.9mmol)和30mln,n-二甲基甲酰胺,室温搅拌溶解,再加入14.5ml3n盐酸(43.5mmol),搅拌过程中有大量固体析出,再加入200ml丙酮。降温至0℃,继续搅拌1h。抽滤,用丙酮洗涤滤饼,真空干燥,得到白色固体9.35g,收率93.5%,hplc纯度为98.8%,将所得固体样品进行粉末x-射线粉末衍射,检测结果为结晶固体,所得图谱见附图1,图谱数据见表1。
表1实施例1所得lesinurad中间体ii结晶粉末x-射线衍射特征峰数据
将所得结晶样品进行差示扫描量热法(dsc)测试,所得图谱吸热峰起始温度为94.45℃,结束温度102.83℃,峰值100.28℃(见附图4)。
实施例2:lesinurad中间体ii结晶的制备
100ml三口瓶中加10glesinurad中间体ii粗品(23.9mmol)和50ml乙醇,加热到30℃,搅拌溶解,再加入3.8ml乙酸(66.3mmol),搅拌过程中有大量固体析出,再加入150ml异丙醇,降温到0℃,继续搅拌2h。抽滤,用异丙醇洗涤滤饼,真空干燥,得到白色固体9.09g,收率90.9%,hplc纯度为98.2%,将所得固体样品进行粉末x-射线粉末衍射,检测结果为结晶固体,所得图谱见附图2,图谱数据见表2。
表2实施例2所得lesinurad中间体ii结晶粉末x-射线衍射特征峰数据
将所得结晶样品进行差示扫描量热法(dsc)测试,所得图谱吸热峰起始温度为93.99℃,结束温度102.53℃,峰值99.90℃(见附图5)。
实施例3:lesinurad中间体ii结晶的制备
500ml三口瓶中加10glesinurad中间体ii粗品(23.9mmol)和200ml乙腈,加热到30℃,搅拌溶解,再加入23.9ml2n硫酸(47.8mmol),搅拌过程中有大量固体析出,降温到0℃,继续搅拌2h。抽滤,用乙腈洗涤滤饼,真空干燥,得到白色固体8.86g,收率88.6%,hplc纯度为98.6%,将所得固体样品进行粉末x-射线粉末衍射,检测结果为结晶固体,所得图谱见附图3,图谱数据见表3。
表3实施例3所得lesinurad中间体ii结晶粉末x-射线衍射特征峰数据
将所得结晶样品进行差示扫描量热法(dsc)测试,所得图谱吸热峰起始温度为95.12℃,结束温度102.56℃,峰值99.91℃(见附图6)。
实施例4:lesinurad的制备(参照wo2014008295方法)
在100ml三口瓶中,加5.1glesinurad中间体ii(实施例1样品,12.2mmol)、25ml甲苯、0.8g氢氧化钠(20mmol)和20ml纯化水,在20℃搅拌反应2h,反应完全。分相,用20ml纯化水萃取有机相,合并水相。浓缩水相至25ml左右,在0~5℃搅拌2h,抽滤,冷水洗涤,真空干燥,得到3.82g白色固体,hplc纯度99.2%。
以下对比例是发明人参照现有技术方法,和/或结合常规方法进行的。按照现有技术方法所得到的lesinurad中间体ii为非固体或无定形固体,纯度差,并且经多种溶剂系统重结晶纯化,所得产品均为油状物,无法得到固体,纯度也未提高。以现有技术方法所得lesinurad中间体ii为原料直接进行lesinurad的制备,所得lesinurad纯度较差,增加了终产品纯化难度。
对比例1:lesinurad中间体ii的制备(参照wo2009070740)
在5l三口瓶中,加71glesinurad中间体iii(0.2mol)、136.7g三乙基苄基氯化铵(0.6mol)和1l溴仿,搅拌溶解,向反应瓶里加138g亚硝酸钠(2mol)和33ml二氯乙酸(0.4mol),室温搅拌反应3h。反应液直接用硅胶柱层析纯化,先用二氯甲烷洗脱,直至溴仿完全洗脱出来,然后用丙酮/二氯甲烷(5:95)洗脱。浓缩洗脱液,得到70g中间体ii,为泡沫状浅黄色固体,收率83.7%,纯度95.3%,xrd检测为无定形固体。
取所得固体样品用自丙酮、乙酸乙酯、丁酮、四氢呋喃、乙腈、二甲亚砜、n,n-二甲基甲酰胺、甲醇、乙醇、异丙醇、正己烷、正庚烷、石油醚等中的一种或多种溶剂进行重结晶,均为油状物,无法得到固体,纯度也未提高。
对比例2:lesinurad的制备(参照wo2014008295)
①lesinurad中间体ii的制备
在200ml四口瓶中,加10glesinurad中间体iv(29.5mmol)和60ml四氢呋喃,35℃下搅拌溶解。降温至30℃,加入7.35gn-溴代丁二酰亚胺(41.3mmol),搅拌反应,hplc监测中间体iv降至0.2%以下(若n-溴代丁二酰亚胺不够,可适当补加)。
降温至5℃,反应液用50ml甲苯和50ml水萃取,分相。然后甲苯相用50ml2%的亚硫酸氢钠溶液洗涤。20℃下,甲苯相分别用50ml6.5%的碳酸氢钠溶液和50ml水洗涤。有 机相一分为二,其中一份有机相(编号中间体ii-1)进行下步反应,另一份(编号中间体ii-2)减压浓缩,得到5.5g黄色粘稠物,纯度91.2%。
②lesinurad的制备
向中间体ii-1的甲苯溶液中加0.8g氢氧化钠(20mmol)和20ml纯化水,在20℃搅拌反应2h,反应完全。分相,用20ml纯化水萃取有机相,合并水相。浓缩水相至25ml左右,在0~5℃搅拌2h,抽滤,冷水洗涤,真空干燥,得到3.75g白色固体,hplc纯度95.7%。
对比例3:lesinurad的制备(参照wo2014008295)
①lesinurad中间体ii的制备
在100ml三口瓶中,加0.71glesinurad中间体iii(2mmol)、1.787g溴化铜(8mmol)和20ml乙腈,控温15~20℃,搅拌下加入3.41g亚硝酸钾(40mol),搅拌直至反应完全。向反应瓶里加24ml3nnaoh溶液和4.61g柠檬酸,搅拌5分钟,再加20ml甲苯萃取,分相。有机相分别用10ml1n氨水和10ml1n柠檬酸钠溶液洗涤。有机相一分为二,其中一份有机相(编号中间体ii-3)进行下步反应,另一份(编号中间体ii-4)有机相减压浓缩,得到0.39g黄色粘稠物,纯度85.8%。
②lesinurad的制备
向中间体ii-3的甲苯溶液中加60mg氢氧化钠(1.5mmol)和5ml纯化水,在20℃搅拌反应2h,反应完全。分相,用5ml纯化水萃取有机相,合并水相。浓缩水相至2ml左右,在0~5℃搅拌2h,抽滤,冷水洗涤,真空干燥,得到0.233g白色固体,hplc纯度95.5%。
- 还没有人留言评论。精彩留言会获得点赞!