利用Pcry3Aa启动子构建高表达海藻糖合成酶工程菌的方法与流程
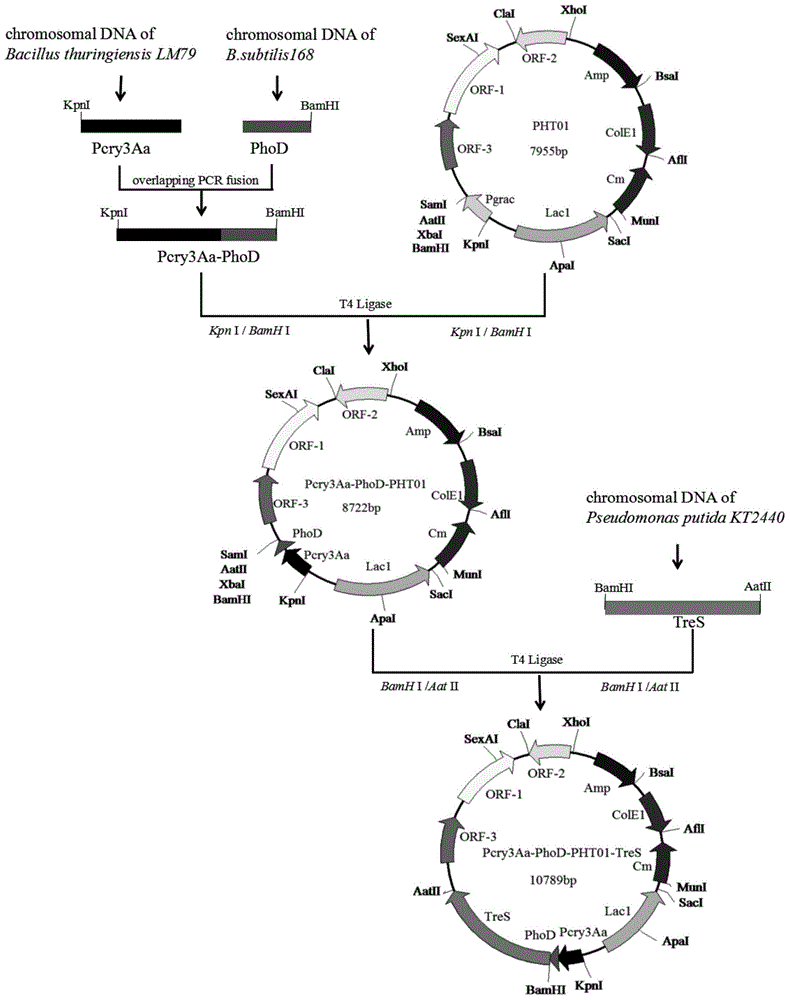
本发明涉及一种利用Pcry3Aa启动子构建高表达海藻糖合成酶工程菌的方法,属于基因工程技术领域。
背景技术:
海藻糖,又称作茧蜜糖、蘑菇糖或蕈糖,不具备还原性,分子式是C12H22O11·2H2O。海藻糖不仅可以储存能量,也可以在特殊情况下起到保护机体的作用。例如在高渗透压、高温、干燥等恶劣条件下,海藻糖可以保护生物体对抗外界的不利因素,来维持生物体的稳定。海藻糖只有在恶劣环境下才会合成,适宜环境下会迅速被降解,这给其生产带来了困难,这也导致其价格较高。因而找到可以大量合成海藻糖的方式是目前需要解决的技术难题。从现阶段来看,海藻糖的制备工艺大体分为四种,分别是微生物抽提法、发酵法、基因重组法和酶转化法。
由于微生物抽提法、发酵法、基因重组法的工艺比较复杂,提取成本较高,制造困难,难以实现规模化生产。酶转化法制备海藻糖是自酶学兴起以来,最具有工业化应用前景的制备方法,其以麦芽糖作底物时,主要是通过TreS实现一步转化。海藻糖酶发现于脂肪杆菌和某些耐热菌中,是目前认为最具有工业化生产潜力的酶。
Pcry3Aa启动子来源于苏云芽孢杆菌(Bacillus thuringiensis,简称Bt),启动表达的cry3Aa蛋白是苏云芽孢杆菌产生的一种对昆虫具有特异毒性的杀虫晶体蛋白,在营养生长阶段表达较弱,然而在进入稳定期的开始阶段被高度激活表达,在稳定期阶段也持续被表达。Pcry3Aa启动子在枯草芽孢杆菌中被营养生长期的sigma因子σA所特异性识别转录,不依赖于枯草芽孢杆菌的稳定期阶段的产芽孢sigma因子以及其它蛋白因子的调控。同时该启动子的激活不要求特别的培养条件、特别的诱导剂和其它压力因素等。因此,Pcry3Aa启动子可作为在枯草芽孢杆菌营养生长期和稳定期表达外源蛋白优选的自诱导启动子之一。
Pcry3Aa启动子启动的转录片段在枯草芽孢杆菌中往往降解成一个小分子的mRNA(5’端在-129处)。小分子mRNA的5’端含有一个特殊的结构STAB-SD(类似于SD序列),该结构与枯草芽孢杆菌的16s rRNA的3’端的结合,决定着整个小分子mRNA的稳定性,延长mRNA的半衰期,从而使得下游的mRNA稳定转录和下游基因的稳定表达,提高了下游基因的表达量。
通过Pcry3Aa启动子所特有的特点用来研究高效表达海藻糖合成酶,进而制备海藻糖具有重要的研究价值。
技术实现要素:
本发明针对现有技术的不足,提供一种利用Pcry3Aa启动子构建高表达海藻糖合成酶工程菌的方法。
本发明技术方案如下:
一种重组载体,其特征在于,在穿梭质粒PHT01的酶切位点BamHI上游插入通过重叠PCR将Pcry3Aa启动子片段和PhoD信号肽片段连接的Pcry3Aa-PhoD片段,在BamHI和AatII两个酶切位点之间插入目的蛋白海藻糖合成酶TreS片段;
所述的Pcry3Aa启动子片段的核苷酸序列如SEQ ID NO.1所示;PhoD信号肽片段的核苷酸序列如SEQ ID NO.2所示;海藻糖合成酶TreS片段的核苷酸序列如SEQ ID NO.3。
上述的能够在枯草芽孢杆菌中高效分泌表达蛋白的Tat型信号肽PhoD片段来源于枯草芽孢杆菌(Bacillus subtilis)的磷酸二酯酶基因信号肽DNA片段。
上述重组载体的制备方法,步骤如下:
(i)提取苏云芽孢杆菌(Bacillus thuringiensis)LM79菌体的基因组,以基因组为模板,进行PCR扩增,得到Pcry3Aa启动子片段;
所述的PCR引物序列如下:
上游引物(Pcry3Aa-F):5’-GGTACCGTGCTTTTTTTGTTGCAATT-3’;
下游引物(Pcry3Aa-R):5’-GACTGTCGTATGCCATTTTTCTTCCTCCCTTTCTTAT-3’;
下划线为酶切位点KpnI;
所述的PCR扩增体系如下:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,51℃退火30sec,72℃延伸40sec,30个循环;72℃延伸10min,-20℃保存;
(ii)提取枯草芽孢杆菌(B.subtilis)168菌体的基因组,以基因组为模板,进行PCR扩增,得到PhoD信号肽片段;
所述的PCR引物序列如下:
上游引物(PhoD-F):
5’-AGGGAGGAAGAAAAATGGCATACGACAGTCGTTTTGATGAATGGG-3’;
下游引物(PhoD-R):5’-GGATCCTACTTCAAAGGCCCCAACCGACTG-3’;
下划线为酶切位点BamHI;
所述的PCR扩增体系如下:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,62℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,-20℃保存;
(iii)将步骤(i)制得的Pcry3Aa启动子片段与步骤(ii)制得的PhoD信号肽片段通过重叠PCR技术连接,制得Pcry3Aa-PhoD片段;
所述重叠PCR扩增体系如下,总体系50μl:
初次扩增体系为25μl:Pcry3Aa启动子片段4μl;PhoD信号肽片段4μl;2×Taq PCR MasterMix 12.5μl;ddH2O 4.5μl;
初次扩增程序如下:
95℃预变性5min;94℃变性30sec,60℃退火30sec,72℃延伸1min,5个循环;
补充扩增体系为25μl:浓度10μmol/L的上游引物Pcry3Aa-F 2μl;浓度10μmol/L的下游引物PhoD-R 2μl;2×Taq PCR MasterMix 12.5μl;ddH2O 8.5μl;
所述的重叠PCR的补充扩增程序如下:
95℃预变性5min;95℃变性30sec,58℃退火30sec,72℃延伸1min,30个循环;72℃延伸10min,-20℃保存;
(iv)将步骤(iii)制得的Pcry3Aa-PhoD片段连接到pTOPO-T vector上,制得pTOPO-T-Pcry3Aa-PhoD质粒载体;然后用限制性内切酶KpnI和BamHI对pTOPO-T-Pcry3Aa-PhoD质粒载体和PHT01质粒进行双酶切,然后采用T4连接酶连接,制得重组质粒Pcry3Aa-PhoD-PHT01;
(v)以恶臭假单胞杆菌的基因组为模板,经PCR扩增得到海藻糖合成酶基因片段TreS;
所述的PCR引物序列如下:
上游引物(TreS-F):5’-CGCGGATCCATGACCCAGCCCGACC-3’
下游引物(TreS-R):5’-CCCAAAGACGTCTCAAACATGCCCGCTGC-3’
单下划线为酶切位点为BamHI,双下划线为酶切位点AatII;
所述的PCR扩增体系如下,总体系50μl:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,54℃退火30sec,72℃延伸2min10sec,30个循环;72℃延伸10min,-20℃保存;
(vi)将步骤(v)制得的酶基因片段TreS连接到pTOPO-T vector上,制得pTOPO-T-TreS质粒载体;然后用限制性内切酶BamHI和AatII对pTOPO-T-TreS质粒载体和重组质粒Pcry3Aa-PhoD-PHT01进行双酶切,然后采用T4连接酶连接,制得重组质粒Pcry3Aa-PhoD-PHT01-TreS。
根据本发明优选的,所述步骤(iv)中,Pcry3Aa-PhoD片段连接到pTOPO-T vector的连接体系如下,总体系10μl:
Pcry3Aa-PhoD片段4μl;pTOPO-T vector 1μl;10×Enhancer 1μl;ddH2O 4μl;
反应条件:室温连接5min;
根据本发明优选的,所述步骤(iv)中,pTOPO-T-Pcry3Aa-PhoD质粒载体双酶切体系如下,总体系40μl:
KpnI内切酶2μl;BamHI内切酶2μl;10×M loading buffer 4μl;pTOPO-T-Pcry3Aa-PhoD 20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
PHT01质粒双酶切体系如下,总体系40μl:
KpnI内切酶2μl;BamHI内切酶2μl;10×M loading buffer 4μl;PHT01质粒20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
所述T4连接酶连接体系如下,总体系10μl:
T4连接酶1μl;10×T4buffer 1μl;双酶切后Pcry3Aa-PhoD基因片段7μl;双酶切后PHT01质粒片段1μl;
反应条件:16℃连接过夜。
根据本发明优选的,所述步骤(v)中的恶臭假单胞杆菌为恶臭假单胞杆菌(Pseudomonas putida)KT2440。
根据本发明优选的,所述步骤(vi)中,所述酶基因片段TreS连接到pTOPO-T vector的连接体系如下,总体系10μl:
TreS片段4μl;pTOPO-T vector 1μl;10×Enhancer 1μl;ddH2O 4μl;
反应条件:混合均匀,室温连接5min;
所述pTOPO-T-TreS质粒载体双酶切体系如下,总体系40μl:
BamHI内切酶2μl;AatII内切酶2μl;10×M loading buffer 4μl;pTOPO-T-TreS质粒载体20μl;ddH2O 12μl;
所述重组质粒Pcry3Aa-PhoD-PHT01双酶切体系如下,总体系40μl:
BamHI内切酶2μl;AatII内切酶2μl;10×M loading buffer 4μl;Pcry3Aa-PhoD-PHT01质粒载体20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
所述T4连接酶连接体系如下,总体系10μl:
T4连接酶1μl;10×T4buffer 1μl;双酶切后TreS基因片段7μl;双酶切后Pcry3Aa-PhoD-PHT01质粒片段1μl;
反应条件:16℃连接过夜。
一种利用Pcry3Aa启动子构建高表达海藻糖合成酶工程菌的方法,包括如下步骤:
(1)以枯草芽孢杆菌168基因组的DNA为模板,PCR扩增获得片段SpoIIID-1;所述SpoIIID-1基因片段核苷酸序列如SEQ ID NO.4所示;
(2)以质粒pBRNeo为模板,PCR扩增卡那霉素抗性基因Kmr片段,制得Kmr片段;所述卡那霉素抗性基因Kmr片段核苷酸序列如SEQ ID NO.5所示;
(3)通过重叠PCR扩增技术,将步骤(1)制得的片段SpoIIID-1与步骤(2)制得的卡那霉素抗性基因Kmr片段进行重叠PCR,扩增得到SpoIIID-1-Kmr片段;
(4)将步骤(3)制得的SpoIIID-1-Kmr片段经BamHI内切酶酶切后转化到枯草芽孢杆菌WB800n中,筛选有卡那霉素抗性的克隆转化子,制得sigK基因不表达的枯草芽孢杆菌WB800n[ΔSpoIIID]菌株;
(5)将上述重组质粒Pcry3Aa-PhoD-PHT01-TreS转化步骤(4)制得的重组枯草芽孢杆菌WB800n[ΔSpoIIID]菌株,筛选有氯霉素抗性的克隆转化子,制得高表达海藻糖合成酶工程菌WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]菌株。
根据本发明优选的,所述步骤(1)中,所述片段SpoIIID-1的PCR扩增引物序列如下:
上游引物(SpoIIID-1-F):5’-CGCGGATCCGCGGGCGCTTTGGTTGATTTGAT-3’;
下游引物(SpoIIID-1-R):
5’-TCTTGTTCAATCATTGGAAACACCAAATTCCTTCGCAATGAC-3’;
单下划线为BamHI酶切位点;片段SpoIIID-1为SpoIIID基因前100bp和SpoIIID基因上游的400bp的整个片段;
所述片段SpoIIID-1的PCR扩增体系为50μl:
2×Taq PCR MasterMix 25μl,浓度为10μmol/L的上游引物2.5μl,浓度为10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,60℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,-20℃保存。
根据本发明优选的,所述步骤(2)中,所述卡那霉素抗性基因Kmr片段的PCR扩增引物序列如下:
上游引物(Kmr-F):
5’-GAATTTGGTGTTTCCAATGATTGAACAAGATGGATTGCACG-3’;
下游引物(Kmr-R):5’-CGCGGATCCTCAGAAGAACTCGTCAAGAAGGCGAT-3’;
单下划线为BamHI酶切位点;
所述卡那霉素抗性基因Kmr片段PCR扩增体系为50μl:
2×Taq PCR MasterMix 25μl,浓度为10μmol/L的上游引物2.5μl,浓度为10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,59℃退火30sec,72℃延伸40sec,30个循环;72℃延伸10min,-20℃保存;
根据本发明优选的,所述步骤(3)中,SpoIIID-1-Kmr片段片段的重叠PCR扩增体系如下,总体系50μl;
初次扩增体系为25μl:片段SpoIIID-1 4μl;卡那霉素抗性基因Kmr片段4μl;2×Taq PCR MasterMix 12.5μl;ddH2O 4.5μl;
初次扩增程序如下:
95℃预变性5min;94℃变性30sec,60℃退火30sec,72℃延伸1min10sec,5个循环;
补充扩增体系为25μl:浓度为10μmol/L的上游引物SpoIIID-1-F 2μl;浓度为10μmol/L的下游引物Kmr-R 2μl;2×Taq PCR MasterMix 12.5μl;ddH2O 8.5μl;
所述的重叠PCR的补充扩增程序如下:
95℃预变性5min;95℃变性30sec,59℃退火30sec,72℃延伸1min10sec,30个循环;72℃延伸10min,-20℃保存。
根据本发明优选的,所述步骤(4)中,SpoIIID-1-Kmr片段转化枯草芽孢杆菌WB800n的步骤如下:
先通过限制性内切酶BamHI酶切SpoIIID-1-Kmr片段,然后将酶切后的片段在1500V、25uF的电击条件下转化到枯草芽孢杆菌WB800n感受态细胞,卡那霉素筛选,即得;
进一步优选的,所述BamHI酶切体系如下,总体系20μl:
BamHI内切酶1μl;10×M loading buffer 2μl;SpoIIID-1-Kmr片段10μl;ddH2O 7μl;
反应条件:30℃恒温4h酶切。
根据本发明优选的,所述步骤(5)中,重组质粒Pcry3Aa-PhoD-PHT01-TreS转化到重组枯草芽孢杆菌WB800n[ΔSpoIIID]的步骤如下:
将重组质粒在1500V、25uF的电击条件下转化到重组枯草芽孢杆菌WB800n[ΔSpoIIID]感受态细胞中,氯霉素筛选,即得。
所述步骤(5)中筛选有氯霉素抗性的克隆转化子的步骤为本领域常规技术,具体如下:
在含有抗生素平板上进行白斑筛选,挑取白色单一斑点,接种到含有氯霉素的液体LB培养基,培养至对数末期,对能在含有抗生素的LB培养基上生长的菌液进行PCR验证,将能够扩增出目的条带的转化子进行提取质粒,对提取的质粒进行酶切验证,得到目的条带。
利用上述制备方法制得的高表达海藻糖合成酶工程菌。
上述高表达海藻糖合成酶工程菌在制备海藻糖合成酶中的应用。
原理说明
本发明的关键技术点是插入来自于苏云芽孢杆菌的Pcry3Aa启动子,该启动子在枯草芽孢杆菌中被营养生长时期以及稳定期的大量存在的E-σA因子识别并结合启动转录,启动子的激活不要求特别的培养条件、特别的诱导剂和其它压力因素等。同时Pcry3Aa启动子转录的mRNA在非翻译区-129位置端含有一个稳定的结构STAB-SD序列,与枯草芽孢杆菌的16s rRNA的结合使得该稳定序列下游的mRNA稳定翻译表达,同时延长了mRNA的半衰期,促使该启动子在枯草芽孢杆菌高强度表达目的蛋白。该技术将Pcry3Aa启动子和信号肽PhoD构建到大肠-枯草芽孢杆菌穿梭质粒PHT01的BamHI酶切位点的上游,在BamHI和AatII两个酶切位点之间插入目的蛋白海藻糖合成酶TreS,成功构建在枯草芽孢杆菌中分泌表达的Pcry3Aa-PhoD-PHT01-TreS重组质粒。
此外,为了提高稳定时期启动子Pcry3Aa更高效的启动转录,通过基因敲出的手段将对sigma因子σA产生负调控的sigK因子进行抑制表达,sigK基因的转录需要SpoIIID的存在,因此,在本发明中通过单交叉互换的形式将SpoIIID-1基因片段与卡那霉素Kmr抗性基因通过重叠PCR连接成片段SpoIIID-1-Kmr。通过电转化将片段SpoIIID-Kmr转化到枯草芽孢杆菌WB800n菌株,得到SpoIIID不表达的WB800n[ΔSpoIIID],然后将重组质粒Pcry3Aa-PhoD-PHT01-TreS转化到WB800n[ΔSpoIIID],得到海藻糖合成酶在营养生长期和稳定期都高效分泌的枯草芽孢杆菌WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]菌株。
有益效果
1、本发明采用苏云金芽孢杆菌的Pcry3Aa启动子自然诱导海藻糖合成酶合成效果显著优于其它诱导剂诱导型表达的效果;
2、本发明的Pcry3Aa启动子含有STAB-SD特殊的结构,该结构与枯草芽孢杆菌16s rRNA序列的结合,提高了其转录mRNA的稳定性以及延长mRNA的半衰期,提高了下游目的基因mRNA的翻译水平,使得海藻糖合成酶高效表达;
3、本发明Pcry3Aa启动子的激活在枯草芽孢杆菌中被营养生长期和稳定时期的sigma因子σA所特异性识别转录,不依赖于枯草芽孢杆菌的稳定期阶段的产芽孢sigma因子以及其它蛋白因子的调控,同时该启动子的激活不要求特别的培养条件、特别的诱导剂和其它压力因素等,符合食品安全级别酶制剂的制备要求,使制得的海藻糖合成酶能够食品医疗行业进行广泛应用;
4、本发明在枯草芽孢杆菌稳定时期,对sigma因子σA产生负调控的sigK因子进行抑制表达,可通过高密度发酵提高海藻糖合成酶的分泌表达产量,显著提高了海藻糖合成酶在稳定时期的分泌表达量,符合工业化制备海藻糖合成酶的需求。
附图说明
图1、Pcry3Aa-PhoD-PHT01-TreS质粒的构建过程示意图;
图2、抑制SpoIIID基因表达过程示意图;
图3、筛选Bacillus subtilis WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]的单菌落实物照片。
具体实施方式
下面结合实施例对本发明的技术方案做进一步阐述,但本发明所保护范围不限于此。
生物材料来源:
枯草芽孢杆菌168(Bacillus subtilis)购自杭州宝赛生物有限公司;
枯草芽孢杆菌WB800n购自杭州宝赛生物有限公司;
恶臭假单胞杆菌(Pseudomonas putida)KT2440购自中国工业微生物菌种保藏中心(CICC);
苏云金芽孢杆菌(Bacillus thuringiensis)LM79购自中国工业微生物菌种保藏中心(CICC);
穿梭质粒PHT01购自杭州宝赛生物有限公司;
pTOPO-T vector购自艾德莱生物科技有限公司;
质粒pBRNeo购自Biovector NTCC保藏中心。
实施例1
将来源于苏云芽孢杆菌的Pcry3Aa启动子与枯草芽孢杆菌的PhoD信号肽通过重叠PCR连接成片段Pcry3Aa-PhoD,将其构建到穿梭质粒PHT01得到重组质粒Pcry3Aa-PhoD-PHT01。
(i)提取苏云芽孢杆菌(Bacillus thuringiensis)LM79菌体的基因组,以基因组为模板,进行PCR扩增,得到Pcry3Aa启动子片段;
所述的PCR引物序列如下:
上游引物(Pcry3Aa-F):5’-GGTACCGTGCTTTTTTTGTTGCAATT-3’;
下游引物(Pcry3Aa-R):5’-GACTGTCGTATGCCATTTTTCTTCCTCCCTTTCTTAT-3’;
下划线为酶切位点KpnI;
所述的PCR扩增体系如下:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,51℃退火30sec,72℃延伸40sec,30个循环;72℃延伸10min,-20℃保存;
经检测,制得的Pcry3Aa启动子片段的核苷酸序列如SEQ ID NO.1所示;
(ii)提取枯草芽孢杆菌(B.subtilis)168菌体的基因组,以基因组为模板,进行PCR扩增,得到PhoD信号肽片段;
所述的PCR引物序列如下:
上游引物(PhoD-F):
5’-AGGGAGGAAGAAAAATGGCATACGACAGTCGTTTTGATGAATGGG-3’;
下游引物(PhoD-R):5’-GGATCCTACTTCAAAGGCCCCAACCGACTG-3’;
下划线为酶切位点BamHI;
所述的PCR扩增体系如下:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,62℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,-20℃保存;
经检测,制得的PhoD信号肽片段的核苷酸序列如SEQ ID NO.2所示;
(iii)将步骤(i)制得的Pcry3Aa启动子片段与步骤(ii)制得的PhoD信号肽片段通过重叠PCR技术连接,制得Pcry3Aa-PhoD片段;
所述重叠PCR扩增体系如下,总体系50μl:
初次扩增体系为25μl:Pcry3Aa启动子片段4μl;PhoD信号肽片段4μl;2×Taq PCR MasterMix 12.5μl;ddH2O 4.5μl;
初次扩增程序如下:
95℃预变性5min;94℃变性30sec,60℃退火30sec,72℃延伸1min,5个循环;
补充扩增体系为25μl:浓度10μmol/L的上游引物Pcry3Aa-F 2μl;浓度10μmol/L的下游引物PhoD-R 2μl;2×Taq PCR MasterMix 12.5μl;ddH2O 8.5μl;
所述的重叠PCR的补充扩增程序如下:
95℃预变性5min;95℃变性30sec,58℃退火30sec,72℃延伸1min,30个循环;72℃延伸10min,-20℃保存;
(iv)将步骤(iii)制得的Pcry3Aa-PhoD片段连接到pTOPO-T vector上,制得pTOPO-T-Pcry3Aa-PhoD质粒载体;然后用限制性内切酶KpnI和BamHI对pTOPO-T-Pcry3Aa-PhoD质粒载体和PHT01质粒进行双酶切,然后采用T4连接酶连接,制得重组质粒Pcry3Aa-PhoD-PHT01;
所述Pcry3Aa-PhoD片段连接到pTOPO-T vector的连接体系如下,总体系10μl:
Pcry3Aa-PhoD片段4μl;pTOPO-T vector 1μl;10×Enhancer 1μl;ddH2O 4μl;
反应条件:室温连接5min;
所述pTOPO-T-Pcry3Aa-PhoD质粒载体双酶切体系如下,总体系40μl:
KpnI内切酶2μl;BamHI内切酶2μl;10×M loading buffer 4μl;pTOPO-T-Pcry3Aa-PhoD 20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
PHT01质粒双酶切体系如下,总体系40μl:
KpnI内切酶2μl;BamHI内切酶2μl;10×M loading buffer 4μl;PHT01质粒20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
所述T4连接酶连接体系如下,总体系10μl:
T4连接酶1μl;10×T4buffer 1μl;双酶切后Pcry3Aa-PhoD基因片段7μl;双酶切后PHT01质粒片段1μl;
反应条件:16℃连接过夜。
(v)以恶臭假单胞杆菌(Pseudomonas putida)KT2440的基因组为模板,经PCR扩增得到海藻糖合成酶基因片段TreS;
所述的PCR引物序列如下:
上游引物(TreS-F):5’-CGCGGATCCATGACCCAGCCCGACC-3’
下游引物(TreS-R):5’-CCCAAATCAAACATGCCCGCTGC-3’
单下划线为酶切位点为BamHI,双下划线为酶切位点AatII;
所述的PCR扩增体系如下,总体系50μl:
2×Taq PCR MasterMix 25μl,浓度10μmol/L的上游引物2.5μl,浓度10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,54℃退火30sec,72℃延伸2min10sec,30个循环;72℃延伸10min,-20℃保存;
经检测,海藻糖合成酶TreS片段的核苷酸序列如SEQ ID NO.3所示;
(vi)将步骤(v)制得的酶基因片段TreS连接到pTOPO-T vector上,制得pTOPO-T-TreS质粒载体;然后用限制性内切酶BamHI和AatII对pTOPO-T-TreS质粒载体和重组质粒Pcry3Aa-PhoD-PHT01进行双酶切,然后采用T4连接酶连接,制得重组质粒Pcry3Aa-PhoD-PHT01-TreS。
所述酶基因片段TreS连接到pTOPO-T vector的连接体系如下,总体系10μl:
TreS片段4μl;pTOPO-T vector 1μl;10×Enhancer 1μl;ddH2O 4μl;
反应条件:混合均匀,室温连接5min;
所述pTOPO-T-TreS质粒载体双酶切体系如下,总体系40μl:
BamHI内切酶2μl;AatII内切酶2μl;10×M loading buffer 4μl;pTOPO-T-TreS质粒载体20μl;ddH2O 12μl;
所述重组质粒Pcry3Aa-PhoD-PHT01双酶切体系如下,总体系40μl:
BamHI内切酶2μl;AatII内切酶2μl;10×M loading buffer 4μl;Pcry3Aa-PhoD-PHT01质粒载体20μl;ddH2O 12μl;
反应条件:37℃恒温4h酶切;
所述T4连接酶连接体系如下,总体系10μl:
T4连接酶1μl;10×T4buffer 1μl;双酶切后TreS基因片段7μl;双酶切后Pcry3Aa-PhoD-PHT01质粒片段1μl;
反应条件:16℃连接过夜。
实施例2
通过重叠PCR技术将来源于枯草芽孢杆菌168的基因片段SpoIIID-1与来自质粒pBRNeo的Kmr连接成片段SpoIIID-1-Kmr,然后通过BamHI限制酶酶切后将片段电转化到枯草芽孢杆菌WB800n,得到SpoIIID基因不表达的菌株Bacillus subtilis WB800n[ΔSpoIIID]。
(1)以枯草芽孢杆菌168基因组的DNA为模板,PCR扩增获得片段SpoIIID-1;所述SpoIIID-1基因片段核苷酸序列如SEQ ID NO.4所示;
所述片段SpoIIID-1的PCR扩增引物序列如下:
上游引物(SpoIIID-1-F):5’-CGCGGATCCGCGGGCGCTTTGGTTGATTTGAT-3’;
下游引物(SpoIIID-1-R):
5’-TCTTGTTCAATCATTGGAAACACCAAATTCCTTCGCAATGAC-3’;
单下划线为BamHI酶切位点;片段SpoIIID-1为SpoIIID基因前100bp和SpoIIID基因上游的400bp的整个片段;
所述片段SpoIIID-1的PCR扩增体系为50μl:
2×Taq PCR MasterMix 25μl,浓度为10μmol/L的上游引物2.5μl,浓度为10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,60℃退火30sec,72℃延伸30sec,30个循环;72℃延伸10min,-20℃保存。
(2)以质粒pBRNeo为模板,PCR扩增卡那霉素抗性基因Kmr片段,制得Kmr片段;所述卡那霉素抗性基因Kmr片段核苷酸序列如SEQ ID NO.5所示;
所述卡那霉素抗性基因Kmr片段的PCR扩增引物序列如下:
上游引物(Kmr-F):
5’-GAATTTGGTGTTTCCAATGATTGAACAAGATGGATTGCACG-3’;
下游引物(Kmr-R):5’-CGCGGATCCTCAGAAGAACTCGTCAAGAAGGCGAT-3’;
单下划线为BamHI酶切位点;
所述卡那霉素抗性基因Kmr片段PCR扩增体系为50μl:
2×Taq PCR MasterMix 25μl,浓度为10μmol/L的上游引物2.5μl,浓度为10μmol/L的下游引物2.5μl,模板2.5μl,用ddH2O补足50μl;
所述的PCR扩增程序如下:
95℃预变性5min;95℃变性30sec,59℃退火30sec,72℃延伸40sec,30个循环;72℃延伸10min,-20℃保存;
(3)通过重叠PCR扩增技术,将步骤(1)制得的片段SpoIIID-1与步骤(2)制得的卡那霉素抗性基因Kmr片段进行重叠PCR,扩增得到SpoIIID-1-Kmr片段;
所述SpoIIID-1-Kmr片段片段的重叠PCR扩增体系如下,总体系50μl;
初次扩增体系为25μl:片段SpoIIID-1 4μl;卡那霉素抗性基因Kmr片段4μl;2×Taq PCR MasterMix 12.5μl;ddH2O 4.5μl;
初次扩增程序如下:
95℃预变性5min;94℃变性30sec,60℃退火30sec,72℃延伸1min10sec,5个循环;
补充扩增体系为25μl:浓度为10μmol/L的上游引物SpoIIID-1-F 2μl;浓度为10μmol/L的下游引物Kmr-R 2μl;2×Taq PCR MasterMix 12.5μl;ddH2O 8.5μl;
所述的重叠PCR的补充扩增程序如下:
95℃预变性5min;95℃变性30sec,59℃退火30sec,72℃延伸1min10sec,30个循环;72℃延伸10min,-20℃保存。
(4)将步骤(3)制得的SpoIIID-1-Kmr片段经BamHI内切酶酶切后转化到枯草芽孢杆菌WB800n中,筛选有卡那霉素抗性的克隆转化子,制得sigK基因不表达的枯草芽孢杆菌WB800n[ΔSpoIIID]菌株;
SpoIIID-1-Kmr片段转化枯草芽孢杆菌WB800n的步骤如下:
先通过限制性内切酶BamHI酶切SpoIIID-1-Kmr片段,然后将酶切后的片段在1500V、25uF的电击条件下转化到枯草芽孢杆菌WB800n感受态细胞,卡那霉素筛选,即得;
所述BamHI酶切体系如下,总体系20μl:
BamHI内切酶1μl;10×M loading buffer 2μl;SpoIIID-1-Kmr片段10μl;ddH2O 7μl;
反应条件:30℃恒温4h酶切。
所述卡那霉素筛选的步骤如下:
在含有抗生素平板上进行白斑筛选,挑取白色单一斑点,接种到含有卡那霉素的液体LB培养基,培养至对数末期,对能在含有抗生素的LB培养基上生长的菌液进行PCR验证,将能够扩增出目的条带的转化子进行提取质粒,对提取的质粒进行酶切验证,得到目的条带。
含有抗生素平板(100ml)组分如下:
蛋白胨1g,酵母粉0.5g,NaCl 1g,琼脂粉2g,250ug卡那霉素;
(5)将实施例1制得的重组质粒Pcry3Aa-PhoD-PHT01-TreS转化步骤(4)制得的重组枯草芽孢杆菌WB800n[ΔSpoIIID]菌株,筛选有氯霉素抗性的克隆转化子,制得高表达海藻糖合成酶工程菌WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]菌株。
所述重组质粒Pcry3Aa-PhoD-PHT01-TreS转化到重组枯草芽孢杆菌WB800n[ΔSpoIIID]的步骤如下:
将重组质粒在1500V、25uF的电击条件下转化到重组枯草芽孢杆菌WB800n[ΔSpoIIID]感受态细胞中,氯霉素筛选,即得。
所述筛选有氯霉素抗性的克隆转化子的步骤,具体如下:
在含有抗生素平板上进行白斑筛选,挑取白色单一斑点,接种到含有氯霉素的液体LB培养基,培养至对数末期,对能在含有抗生素的LB培养基上生长的菌液进行PCR验证,将能够扩增出目的条带的转化子进行提取质粒,对提取的质粒进行酶切验证,得到目的条带。
含有抗生素平板(100ml)组分如下:
蛋白胨1g,酵母粉0.5g,NaCl 1g,琼脂粉2g,250ug氯霉素。
实施例3
海藻糖合成酶的生产,操作步骤如下:
1)一级种的制备:从含氯霉素20ug/mL的LB平板上转接实施例2制得的高表达海藻糖合成酶工程菌WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]菌株单菌落在3mL LB液体培养基37℃、200rpm培养过夜,所得的菌种为一级种;
2)二级种的制备:一级种接种于含氯霉素20ug/mL的800mL LB液体培养基中,于37℃、200rpm培养至OD600为0.9(4.5小时);
3)三级种的制备:将二级种接种到80L LB液体发酵罐中,37℃,以柠檬酸、NaOH控制pH7.0左右,通风搅拌,溶氧控制在20-30%,培养至OD600为0.9(4.5小时);
4)生产罐发酵:将三级种接种到3吨发酵罐中,LB液体培养基,36~37℃,通风搅拌,溶氧控制在20~30%,以柠檬酸、NaOH控制pH6~8,培养35~38小时,10000g离心除菌,用截留分子量5000~10000超滤膜浓缩上清液后,制得海藻糖合成酶浓缩原液;
5)海藻糖合成酶酶活的测定按如下方法进行:
取1mL制得的海藻糖合成酶浓缩原液与1mL质量浓度为60%的麦芽糖溶液(用50mM pH6.5磷酸缓冲液配制)混合,混匀后在37℃反应30min,反应液10000rpm离心3min取上清。
用按还原糖测定方法(3,5-二硝基水杨酸比色法)测定还原糖含量。
对照样取1ml质量浓度为60%的麦芽糖溶液与1ml 50mM pH6.5磷酸缓冲液混合,37℃反应30min,10000rpm离心3min取上清,同上方法测定还原糖含量。
活力单位(U)定义为1ml酶液在pH6.5、37℃条件下,每分钟转化1μg麦芽糖为非还原糖为一个海藻糖合成酶活力单位。计算方法如下:
U(μg/ml·min)=(V1×M×(OD1-OD2)/OD1)/(T×V2)=(1ml×100mg/ml×(OD1-OD2)/OD1×1000)/(30min×1ml)
V1:底物体积(ml),V2:参加反应的酶液体积(ml),M:底物溶度(mg/ml),OD1:对照样测还原糖时的吸光值,OD2:样品测还原糖的吸光值,1000:mg转化成μg的系数,T:酶反应时间(min)。
经测定,发酵原液离心后,各枯草芽孢杆菌基因工程菌株发酵表达的最高酶活(包括发酵液中已经转化完的海藻糖部分)见表1:
表1
结果分析:
Pcry3Aa启动子启动的转录片段在枯草芽孢杆菌中往往降解成一个小分子的mRNA(5’端在-129处)。小分子mRNA的5’端含有一个特殊的结构STAB-SD(类似于SD序列),该结构与枯草芽孢杆菌的16s rRNA的3’端的结合,决定着整个小分子mRNA的稳定性,延长mRNA的半衰期,从而使得下游的mRNA稳定转录和下游基因的稳定表达,提高了下游基因的表达量。
在进入稳定期后,sigma因子σA会一直识别结合启动子表达,然而σA同时受到芽孢生成的调控sigma因子σk的反馈抑制作用,使得σA因子对启动子Pcry3Aa识别结合会被减弱,影响到启动子下游基因的表达量。sigK基因是由spoIVCB和spoIIIC基因通过去除大约42kb的间插DNA的位点特异性重组而得来的。重组需要SpoIVCA编码的重组酶。由sigK基因的转录也需要SpoIIID的存在。因此,该技术选择SpoIIID基因的不表达会大大降低sigK基因的转录水平,σk因子对σA因子的负调控作用消失。
因此,在含有SpoIIID基因不表达的WB800n[ΔSpoIIID]+[Pcry3Aa-PhoD-PHT01-TreS]菌株在稳定期提高了sigma因子σA的表达量,进而提高了σA对启动子Pcry3Aa的识别与结合,提高了海藻糖合成酶的表达量,其表达效果要高于WB800n[Pcry3Aa-PhoD-PHT01-TreS]菌株。
- 还没有人留言评论。精彩留言会获得点赞!