一种乙胺嘧啶的分离检测方法与流程
本发明涉及有机化学和分析化学领域,尤其涉及一种乙胺嘧啶的分离检测方法。
背景技术:
乙胺嘧啶在动物产品和水产品养殖中也有着广泛的应用,适量使用能增强动物的抗病能力,是一种广谱抗菌兽药,主要用于防治鸡球虫病、禽霍乱及仔猪白痢等。但是乙胺嘧啶在水产品体内具有高度的蓄积性,超出一定范围,人食用后破坏人的造血系统,造成溶血性贫血症,甚至有引起潜在致癌性的可能,同时对中枢神经系统有直接的毒性作用。英、美早己明文规定禁用乙胺嘧啶等作饲料添加剂,日本对乙胺嘧啶残留量一律执行标准,但国内尚没有乙胺嘧啶残留的限量标准。因此,针对乙胺嘧啶有致癌的可能性和对神经中枢的毒害作用,特异性检测乙胺嘧啶是十分重要的。
高效液相色谱(HPLC)分析生物样品中的药物,一般使用C18柱,在注射进色谱柱前往往涉及到复杂的前处理,比如通过加酸或者有机溶剂沉淀降解,以及液液萃取和离线固相萃取。但是这些过程复杂而且耗时,药物也可能在样品前处理阶段损失,所以在这样情况下,需要产生一种对目标分子有选择性的材料。分子印迹技术是一种将分子识别位点引入聚合材料的制备技术。分子印迹技术分为分子预组装过程(即共价键结合作用)以及分子自组装过程(即非共价键结合用)。有关研究结果显示,分子印迹聚合物作为固相萃取吸附剂不仅选择性高,而且具有结合力强、可重复利用和成本低的优点。
当前,分子印迹-固相萃取在实际应用中需要以分子印迹聚合物为填料制作固相萃取柱,而现有的分子印迹聚合的反应条件较苛刻,并且反应时间长。这样制作固相萃取柱不仅操作繁琐,而且萃取过程中柱压高、流速低,这些弊端在很大程度上限制了分子印迹-固相萃取的进一步推广与应用。因而研发一种简单易合成的分子印迹聚合物实现对乙胺嘧啶的选择性检测具有重要意义。
技术实现要素:
本发明的目的在于克服现有技术中的缺点和不足,提供一种操作简单的乙胺嘧啶的分离检测方法。
本发明是通过以下技术方案实现的:一种乙胺嘧啶的分离检测方法,包括以下步骤:
(1)样品处理:提取样品中的乙胺嘧啶,作为样品待测液;
(2)向待测液中加入乙胺嘧啶分子印迹聚合物,震荡后离心分离乙胺嘧啶分子印迹聚合物;
(3)用一定体积的洗脱溶剂洗脱乙胺嘧啶分子印迹聚合物中的乙胺嘧啶,收集洗脱液;
(4)用HPLC/UV测定洗脱液中乙胺嘧啶的浓度;
所述步骤(2)中的乙胺嘧啶分子印迹聚合物以乙胺嘧啶为模板,加入功能单体、接枝双键的二氧化硅微球、巯基交联剂,通过巯-烯基点击化学反应制备得到。
相对于现有技术,本发明的乙胺嘧啶的分离检测方法,使用乙胺嘧啶分子印迹聚合物,在所述分子印迹聚合物制备过程中使用含有巯基的交联剂,利用巯烯基点击化学结合分子印迹技术,使得反应条件温和,反应快速。并且通过优化巯烯基点击反应合成分子印迹聚合物的反应条件,得到该反应的最优反应条件,大大地提高了制备得到的分子印迹聚合物对乙胺嘧啶的吸附容量及选择性能。
进一步,所述乙胺嘧啶分子印迹聚合物的功能单体为甲基丙烯酸。
进一步,所述巯基交联剂为季戊四醇四-3-巯基丙酸酯,所述引发剂为三乙胺,三乙胺调节pH=8,引发反应,所述分散剂为甲苯。
进一步,步骤(4)中,所述HPLC/UV的检测条件为:色谱柱为C18柱;流动相为体积比为8:2的甲醇和水混合溶剂;流速为1.0mL/min;UV的检测波长为λ=272nm;检测温度为30℃。
进一步,步骤(3)中,所述洗脱溶剂为甲醇。
进一步,步骤(2)中所述乙胺嘧啶分子印迹聚合物的制备方法包括以下步骤:
S21:合成接枝双键的的二氧化硅微球;
S22:将乙胺嘧啶和功能单体溶解于溶剂中自组装,充分摇匀;
S23:向步骤S22的溶液中加入接枝双键的二氧化硅微球、巯基交联剂、引发剂和分散剂,使其发生聚合反应,得到表面包覆模板分子的分子印迹聚合物;
S24:洗涤步骤S23中得到的表面包覆乙胺嘧啶的分子印迹聚合物,除去其中包覆的乙胺嘧啶后,干燥,得到乙胺嘧啶分子印迹聚合物。
进一步,所述步骤23中,还加入辅助交联剂,所述辅助交联剂为二季戊四醇五烯丙酸。
进一步,所述步骤S23中,乙胺嘧啶、功能单体的摩尔比为1:2;所述巯基交联剂与接枝双键的二氧化硅微球中的双键的摩尔比为1:1。
为了更好地理解和实施,下面结合附图详细说明本发明。
附图说明
图1是在待测样品中加入分子印记聚合物前(A)和后(B)的液相色谱分析,其中1-磺胺嘧啶,2-双酚A,3-氨苯蝶啶,4-乙胺嘧啶。
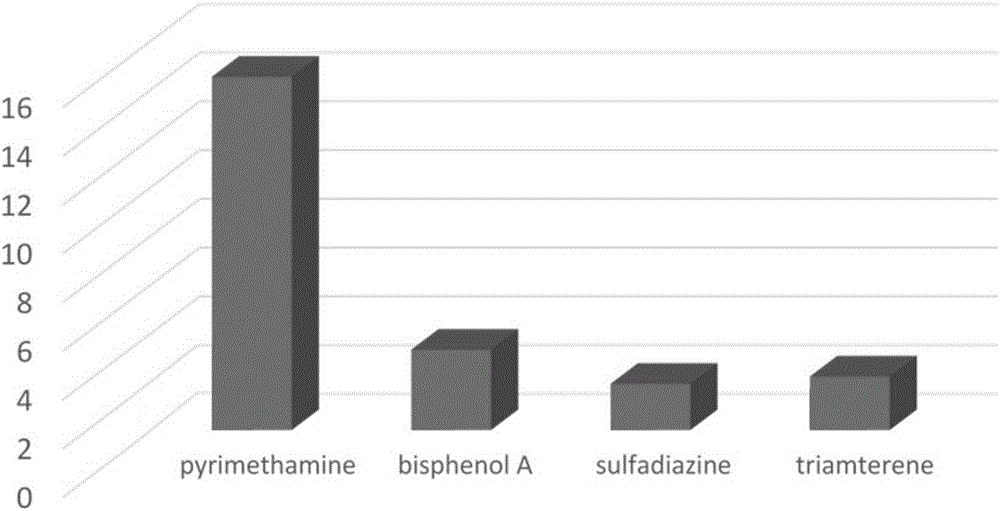
图2是本发明的分子印迹聚合物分别对不同分子的选择性萃取容量图。
具体实施方式
本发明公开的一种乙胺嘧啶的分离检测方法,包括以下步骤:
(1)样品处理:提取样品中的乙胺嘧啶,作为样品待测液。
(2)向待测液中加入乙胺嘧啶分子印迹聚合物,震荡后离心分离乙胺嘧啶分子印迹聚合物。
(3)用一定体积的洗脱溶剂洗脱乙胺嘧啶分子印迹聚合物中的乙胺嘧啶,收集洗脱液。
(4)用HPLC/UV测定洗脱液中乙胺嘧啶的浓度。
在本实施例中,以鱼肉样品为例。取2g鱼肉样品剪碎后匀浆,在匀浆样品中加入4g无水硫酸钠和10mL体积比为5:5的乙腈-三氯甲烷混合液,充分混合后,5000r/min转速离心5min,取有机相,残渣再加10mL体积比为5:5的乙腈-三氯甲烷混合液萃取,将两次有机相合并作为鱼肉样品的待测液。
在步骤(2)中,取3mL的鱼肉样品待测液,向待测液中加入10~50mg的分子印迹聚合物,震荡0.5~4h后,离心分离分子印迹聚合物。在本实施例中,所述分子印迹聚合物为以乙胺嘧啶为模板,加入功能单体、接枝双键的二氧化硅微球、巯基交联剂、引发剂和分散剂,通过巯-烯基点击化学反应制备得到。所述功能单体为甲基丙烯酸,所述巯基交联剂为季戊四醇四-3-巯基丙酸酯。在本实施例中,还可以加入辅助交联剂,所述辅助交联剂为二季戊四醇五烯丙酸。所述模板分子乙胺嘧啶与功能单体甲基丙烯酸的摩尔比为1:1~1:3,优选摩尔比为1:2;所述巯基交联剂与接枝双键的二氧化硅微球中的双键的摩尔比为1:2~2:1,优选摩尔比为1:1。所述引发剂为三乙胺,利用三乙胺调节pH=8,引发反应。
在步骤(3)中,用2mL的洗脱溶剂洗脱除去分子印迹聚合物中所包覆的乙胺嘧啶。重复多次,并收集洗脱液。乙胺嘧啶溶剂于洗脱溶剂中,所述洗脱溶剂为甲醇溶液。
在步骤(4)中,用HPLC/UV测定洗脱液中乙胺嘧啶的浓度。所述色谱条件如下:所述色谱柱为C18柱;流动相为体积比为8:2的甲醇和水混合溶剂;所述流速为1.0mL/min;所述UV的检测波长为λ=272nm;所述检测温度为30℃。
具体的,本实施例提供一种步骤(2)中所述分子印迹聚合物的制备方法,但不局限于此。所述分子印迹聚合物的制备方法如下:
S21:合成接枝双键的的二氧化硅微球;
S22:将模板分子和功能单体溶解于溶剂中自组装,充分摇匀;
S23:向步骤S22的溶液中加入接枝双键的二氧化硅微球、巯基交联剂、引发剂和分散剂,使其发生聚合反应,得到表面包覆模板分子的分子印迹聚合物;
S24:洗涤步骤S23中得到的表面包覆模板分子的分子印迹聚合物,除去其中包覆的模板分子后,干燥,得到分子印迹聚合物;
其中,所述模板分子为乙胺嘧啶,所述功能单体为甲基丙烯酸,所述巯基交联剂为季戊四醇四-3-巯基丙酸酯,所述模板分子乙胺嘧啶与功能单体甲基丙烯酸的摩尔比为1:1~1:3,优选摩尔比为1:2;所述巯基交联剂与接枝双键的二氧化硅微球中的双键的摩尔比为1:2~2:1,优选摩尔比为1:1。在步骤S23中,还可加入辅助交联剂,所述辅助交联剂为二季戊四醇五烯丙酸。
请参阅图1,其是在待测样品中加入分子印迹聚合物前(A)和后(B)的液相色谱分析。从图中可知,在加入分子印迹聚合物之前,通过液相色谱可检测到较强的乙胺嘧啶的峰。而在加入分子印迹聚合物后,通过液相色谱基本检测不到乙胺嘧啶的峰。但其他峰基本保持不变,说明了所述分子印迹聚合物对乙胺嘧啶具有很高的选择吸附性能。配制同时含有氨苯蝶啶、双酚A、磺胺嘧啶、乙胺嘧啶的混合溶液,加入分子印迹聚合物震荡后,取上层清夜进行液相色谱分析。从图中可知,所述分子印迹聚合物对乙胺嘧啶的吸附量远远高于对其他三种类似物的吸附量,说明了所述分子印迹聚合物对乙胺嘧啶表现出良好的选择性吸附性能。产生该结果的原因是在合成所述分子印迹聚合物的过程中加入了乙胺嘧啶而形成了功能单体-模板分子复合物,当洗涤出模板分子后在聚合物内部形成了形状和官能团位置均与乙胺嘧啶相配合的空腔。如果待吸附溶液含有乙胺嘧啶的话,分子印迹聚合物的空腔恰好能容纳乙胺嘧啶,且空腔内的官能基团会与乙胺嘧啶产生氢键作用而结合在一起,形成特异性吸附。
请参阅图2,其是本发明的分子印迹聚合物分别对不同分子的选择性萃取容量图,其中选取的分子依次为乙胺嘧啶(pyrimethamine)、双酚A(bisphenol A)、磺胺嘧啶(sulfadiazine)和氨苯蝶啶(triamterene)。从图中可知,分子印迹聚合物对乙胺嘧啶的萃取量为14.5mg/g,而所述分子印迹聚合物分别对于双酚A、磺胺嘧啶和氨苯蝶啶的萃取量相差不大,且萃取量都较低,说明了相对于其他结构类似的物质,本发明的分子印迹聚合物对乙胺嘧啶具有较好的选择性。
本发明对所述分子印迹聚合物对乙胺嘧啶的分离检测进行了回收实验,不含乙胺嘧啶的鱼肉样品处理后,向其中加入一定浓度的乙胺嘧啶标准溶液,配制成低、中、高浓度的鱼肉加标样品,取一定量的分子印迹聚合物加入其中,混合物密封后放置于摇床中震荡12h,离心分离分子印迹聚合物。用甲醇溶液对分子印迹聚合物进行洗脱,将洗脱液用HPLC/UV检测,求得加标回收率。其中加标回收率=实测浓度/理论浓度,经实验得到所述分子印迹聚合物对乙胺嘧啶的回收率可达90%以上,最低检测限为7.5μg/g,能实现对微量痕量的乙胺嘧啶的检测。本发明所述的分子印迹聚合物可重复使用,并在重复使用多次后仍旧保持很高的对乙胺嘧啶的选择吸附性能。
相对于现有技术,本发明的乙胺嘧啶的分离检测方法,使用乙胺嘧啶分子印迹聚合物,在所述分子印迹聚合物制备过程中使用含有巯基的交联剂,利用巯烯基点击化学结合分子印迹技术,使得反应条件温和,反应快速。并且通过优化巯烯基点击反应合成分子印迹聚合物的反应条件,得到该反应的最优反应条件,大大地提高了制备得到的分子印迹聚合物对乙胺嘧啶的吸附容量及选择性能。
本发明并不局限于上述实施方式,如果对本发明的各种改动或变形不脱离本发明的精神和范围,倘若这些改动和变形属于本发明的权利要求和等同技术范围之内,则本发明也意图包含这些改动和变形。
- 还没有人留言评论。精彩留言会获得点赞!