一种氟苯尼考中间体、及其制备方法和氟苯尼考的制备方法与流程
本发明涉及兽药制备领域,具体涉及制备氟苯尼考的中间体和氟苯尼考的方法。
背景技术:
:
氟苯尼考又称氟甲砜霉素,氟砜尼可,是一类化学合成的抗生素,开发用于动物保健市场的新型抗革兰氏阳性菌,是动物专用广谱抗菌药,其结构与甲砜霉素相似,有两个手性中心,四个对应异构体,但只有其中一个有药理活性。
目前国内较成熟的合成氟苯尼考的工艺路线,大多采用对甲砜苯甲醛路线(Tobibiki Hisao;Ger Offen,DE2349496),均以氯霉素、甲砜霉素这些原料药或其相关中间体作为起始原料,经还原,保护,氟代,水解开环,酰化制得。其中涉及到对甲砜苯甲醛与甘氨酸、硫酸铜反应制备铜盐后经酯化反应得到外消旋的D、L型丝氨酸乙酯,经酒石酸的拆分得到,其中一个异构体L型被废弃,是整个路线的成本增加,且制备铜盐时会产生大量的硫酸铜废水,废水处理成本非常高,使得企业因生产成本和环保压力处于临时关停状态。
随着不对称化学的发展,各种手性催化剂开始应用于氟苯尼考的合成。美国的Jon E.Clark等人采用酶催化的方法进行了该类反应的研究,后来Feng Li等人采用不对称合成的方法合成中间体噁唑林,然后再经水解、二氯乙酰化反应制得。该工艺路线存在收率低、流程较长的缺点,很难应用于工业化生产(Tetrahedron:Asymmetry 22(2011)1337-1341)。
技术实现要素:
:
为克服现有技术中的上述问题,本发明提供了一种生产氟苯尼考使用的关键中间体(IV)的制备方法,还提供了氟苯尼考的制备方法,制备出氟苯尼考,成本低,工艺简单,收率高且产品手性纯度高达98%。
一种如式(IV)所示的中间体,其特征在于:所述的中间体结构式为
R1为甲硫醚基或者亚甲砜基或者甲砜基;
R2为TBS-或者TMS-或者MOM-或者THP-;
R3为或者或者
R4为二氯乙酰基或者苯甲酰基或者叔丁氧羰基。
所述的中间体(IV)
R1为:甲硫醚基
R2为:MOM-
R3为:
R4为:二氯乙酰基。
一种如式(IV)所示的中间体的制备方法,其制备方法如下:
(1)加成反应:将取代的苯甲醛(SM)和氰化钾在(S)-羟氰裂解酶的作用下反应,得到中间体(I);
(2)取代反应:中间体(I)对羟基进行保护后得到中间体(II);
(3)加成-还原反应:中间体(II)与R3的格式试剂加成后经硼氢化钠还原,经过重结晶后得到中间体(III);
(4)酰化反应:中间体(III)与酰氯溶解于乙酸乙酯中,在傅酸剂的作用下,进行酰化反应,得到中间体(IV)。
所述的中间体(IV)
R1为:甲硫醚基
R2为:MOM-
R3为:
R4为:二氯乙酰基。
一种氟苯尼考的制备方法,其步骤如下:
(1)加成反应:将取代的苯甲醛(SM)和氰化钾在(S)-羟氰裂解酶的作用下反应,得到中间体(I);
(2)取代反应:中间体(I)对羟基进行保护后得到中间体(II);
(3)加成-还原反应:中间体(II)与R3的格式试剂加成后经硼氢化钠还原,经过重结晶后得到中间体(III);
(4)酰化反应:中间体(III)与酰氯溶解于乙酸乙酯中,在傅酸剂的作用下,进行酰化反应,得到中间体(IV);
(5)氧化反应:中间体(IV)经高锰酸钾在丙酮和水中反应,氧化成酸,得到中间体(V):
(6)还原反应:中间体(V)在低温下经过硼氢化钠和三氟化硼的还原作用,将羧酸还原成羟基,得到中间体(Ⅵ);
(7)氟化反应:中间体(Ⅵ)在进行氟化反应得到中间体(VII);
(8)水解反应:中间体(Ⅶ)在酸的醇溶液中,发生水解即得到氟苯尼考(TM)。4.根所述的R1为甲硫醚基或者亚甲砜基或者甲砜基;
R2为TBS-或者TMS-或者MOM-或者THP-;
R3为或者或者
R4为二氯乙酰基或者苯甲酰基或者叔丁氧羰基。
步骤(3)中R3的溴代物与镁屑在氮气保护下,经过引发之后得到格式试剂,然后与中间体(II)进行加成反应后,再经过硼氢化钠还原得到中间体(III)。
步骤(3)反应得到的中间体进行重结晶,重结晶采用的溶剂为乙酸乙酯和石油醚,体积比例为1∶2-1∶3。
步骤(4)中,中间体(III)和酰氯的摩尔比为1∶1.05,傅酸剂为三乙胺、吡啶、N、N-二异丙基乙胺中的一种。
步骤(5)中,中间体(IV)和高锰酸钾的摩尔比为1∶4-1∶5。
步骤(6)中,中间体(V):硼氢化钠:三氟化硼的摩尔比为1∶2∶2.6,温度-10℃--30℃。
步骤(8)中,所述的酸为卤代酸或者三氟乙酸的一种,所述的醇为:甲醇或者乙醇或者异丙醇。
最佳的实施路径如下:
有益效果:
1、将手性催化的不对称合成放在整个工艺的前期,并且为简单的化学反应,可以节约成本,且催化剂可以回收,重复使用,提高了收率;
2、得到的中间体烯丁胺手性纯度大于98%;
3、整个工艺路线简洁,原料便宜易得,成本低,操作简便,收率高,制备出的氟苯尼考手性纯度高且收率高。
4、改变目前氟苯尼考工业化生产面临的资源浪费、生产成本高、环保压力大的现状,采用手性羟氰酶不对称催化合成羟氰和手性诱导还原得到关键手性中间体(IV),反应选择性强,收率高,工艺简单,成本低,适用于工业化生产;避免了传统工艺常用的手性拆分,节约了资源,减轻环保压力,降低了生产成本。
说明书附图
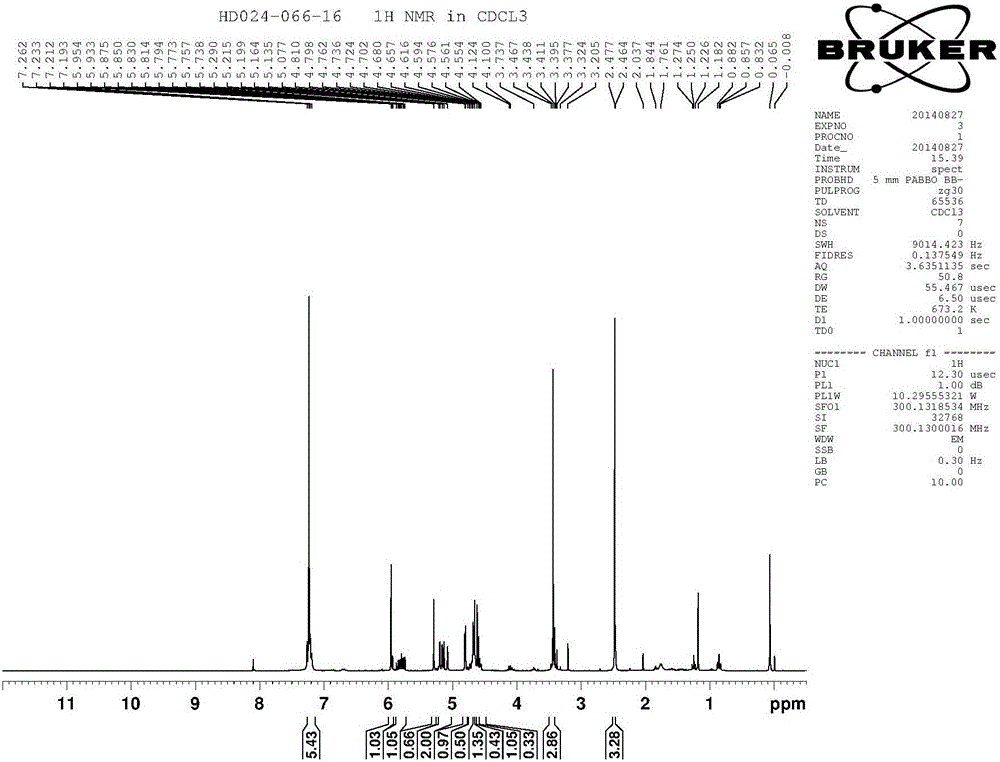
图1为氟苯尼考的氢谱图
图2为化合物(IV)的氢谱图
具体实施方式
实施例I:
将KCN(307g,4.71mol)加入到柠檬酸配制的缓冲溶液中(400mL,PH=5.0),冷却至0℃后,备用。将对甲硫醚苯甲醛(71.6g,0.471mol)溶解于TBME(400mL)后,加入至反应液,维持0℃,加入(S)-HNL(40mL),搅拌1.5小时,HPLC检测反应结束。加入5M HCl(50mL)淬灭反应,催化剂生成沉淀后,过滤,滤液用DCM(3×50mL)萃取,合并有机相,干燥,浓缩得化合物I(83.1g,98.6%)。
1H NMR(CDCl3 300MHz):δ2.5(s,3H),3.0(br,s,1H),5.49(s,1H),7.29-7.43(AB,J=8.5Hz,4H).
实施例II:
将化合物I(10g,55.8mmol)溶于CH3CN(150mL),加入甲缩醛(12.5g,167.4mmol),分批加入P2O5(23.5g,167.4mmol),TLC检测至反应结束。将反应液倒入水中,减压除去乙腈后,加入TBME(100mL×2)萃取,合并有机相,分别用饱和NaHCO3(100ml×1)、饱和NaCl溶液(500mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩得化合物IIa(12.4g,83.2%)。
将对甲硫醚苯甲醛(10g,65.7mmol)溶于THF(70mL)中,加入溴化锌(1.5g,6.57mmol),于氮气保护下降温至0-5℃后,滴加TMSCN(6.5g,6.57mmol),反应25hs,HPLC检测反应结束。将反应液倒入水中,加入TBME(100mL×2)萃取,合并有机相,分别用饱和NaHSO3(100ml×1)、饱和NaCl溶液(500mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩得化合物IIb(12.7g,76.9%)。
将化合物I(2g,11.1mmol)溶于DCM中(15mL),加入二氢吡喃(1.4g,16.7mmol),分批加入PPTS(55.8mg,0.22mmol),TLC检测至反应结束。将反应液倒入水中,减压除去DCM后,加入TBME(10mL×2)萃取,合并有机相,分别用饱和NaHCO3(10ml×1)、饱和NaCl溶液(50mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩得化合物IIc(2.1g,72.4%)。
实施例III:
将化合物IIa(5.0g,22.4mmol)溶于100mL乙醚中,降温至0℃,滴加乙烯基溴化镁(24mL,67.2mmol),在此温度下搅拌1小时,HPLC检测原料消失后加入甲醇(70mL),分批加入硼氢化钠(3.4g,89.6mmol),搅拌1.5小时,HPLC检测反应结束,加入冰水淬灭,TBME(50mL×2)萃取,合并有机相后,经过水洗(50mL×2)、饱和氯化钠(50mL×2)洗后,干燥,浓缩得粗品(6.8g),经过EA/PE体系重结晶得到白色固体化合物Ⅲa(5.4g,95%)。
将1,3-二噻烷(0.48g,3.99mmol)加入乙醚(20mL),冷却至-78℃,滴加正丁基锂(2.5mL,3.99mmol),搅拌30min后,加入化合物IIb(0.5g,1.98mmol)的乙醚溶液,在-78℃下反应1.5hs,加入甲醇(10mL)、硼氢化钠(0.307g,7.99mmol),搅拌1.5小时,HPLC检测反应结束,加入1N HCl(10mL)淬灭,TBME(20mL×2)萃取,合并有机相后,经过水洗(20mL×2)、饱和氯化钠(20mL×2)洗后,干燥,浓缩得化合物Ⅲb(0.45g,60.2%)。
将化合物IIc(2.0g,7.6mmol)溶于100mL乙醚中,降温至0℃,滴加2-噻吩基溴化镁(11.4mL,22.8mmol),在此温度下搅拌1小时,HPLC检测原料消失后加入甲醇(20mL),分批加入硼氢化钠(1.15g,30.4mmol),搅拌1.5小时,HPLC检测反应结束,加入冰水淬灭,TBME(20mL×2)萃取,合并有机相后,经过水洗(20mL×2)、饱和氯化钠(20mL×2)洗后,干燥,浓缩得化合物Ⅲc(1.9g,73.0%)。
实施例IV:
将化合物Ⅲa(5.6g,22.1mmol)加入乙酸乙酯中(60mL),搅拌溶解后,加入三乙胺(2.4g,24.3mmmol),降温至0℃,加入二氯乙酰氯(3.4g,23.2mmol),搅拌1小时,HPLC检测反应结束后,加入水淬灭反应,分层,有机相分别用磷酸二氢钠(50mL×1))、饱和氯化钠(50mL×1))洗涤,无水硫酸钠干燥,抽滤浓缩至干得到固体化合物IVa(8.8g,100%)。
1H NMR(CDCl3 300MHz):δ2.58(s,3H),3.46(s,3H),4.59-4.73(m,1H),4.79-4.81(m,1H),5.07-5.21(m,2H),5.29(s,2H),5.74-5.87(m,1H),5.95(s,1H),7.19-7.26(m,4H).
将化合物Ⅲb(10.0,26.7mmol)加入乙酸乙酯中(100mL),搅拌溶解后,加入N、N-二异丙基乙胺(3.8g,29.4mmmol),降温至0℃,加入苯甲酰酰氯(3.94g,28.0mmol),搅拌1小时,HPLC检测反应结束后,加入水淬灭反应,分层,有机相分别用磷酸二氢钠(100mL×1))、饱和氯化钠(100mL×1))洗涤,无水硫酸钠干燥,抽滤浓缩至干得到固体化合物IVb(11.2g,87.7%)。
实施例V:
将化合物4(10g,27.4mmol)溶于丙酮(60mL)中,加入水(60mL),降温至0℃,滴加高锰酸钾(21.7g,137mmmol)的丙酮(30mL)溶液,滴加结束后,继续搅拌2小时,HPLC检测反应结束,加入3NHCl(10ml)淬灭反应,乙酸乙酯(100mL×2)萃取,合并有机相,分别用磷酸二氢钠(100mL×1)、饱和氯化钠(100mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩至干得到固体化合物5(8.5g,94.4%)。
实施例VI:
将硼氢化钠(3.6g,92.6mmol)加入到干燥的THF中(80mL),冷却至-10℃后,缓慢滴加BF3·Et2O(15.6ml,121mmol),耗时约1小时;滴加结束后,加入化合物5(19.1g,46.3mmol)的THF(60mL)溶液,继续搅拌5小时,HPLC检测反应结束。加入甲醇(20mL)淬灭反应后,继续加入10%HCl(50mL)并加热至60℃,搅拌1小时,冷却至室温,乙酸乙酯(100mL×2)萃取,合并有机相,分别用20%NaOH(100mL×1)、饱和氯化钠(100mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩至干得到固体化合物6(15.9g,86.1%)。
实施例VII:
将化合物6(5.1g,12.8mmol)溶于THF(50mL)中,加入Et3N(3.9g,38.4mmol),Et3N·3HF(3.1g,19.2mmol),控制温度在25℃,滴加PBSF(5.8g,19.2mmol),滴加结束后,室温搅拌12hs。HPLC检测原料反应结束,加入DCM(50mL)和H2O(50mL),分层,水相加入DCM(50mL×2)萃取,合并有机相,用饱和NaCl溶液(100mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩得粗品化合物7(5.1g,100%)。
实施例VIII:
将化合物7(4.5g,12.8mmol)溶于i-PrOH(26mL)和HCl(14mL)中,加热回流3hs,HPLC检测反应结束后,冰浴冷却至室温,加入DCM(50mL×2)萃取,合并有机相,用饱和NaCl溶液(100mL×1)洗涤,无水硫酸钠干燥,抽滤浓缩得粗品,加入甲苯重结晶得到白色晶体目标化合物(4.15g,91%)。
1H NMR(DMSO 300M):63.15(s,3H),4.23-4.29(m,1H),4.30-4.72(m,2H),4.97(d,1H),6.13(d,1H),6.44(s,1H),7.59(d,2H),7.83(d,2H),8.59(d,1H)。
- 还没有人留言评论。精彩留言会获得点赞!