一种杂化抗癌药物及其制备方法与应用与流程
本发明属于医药技术领域,特别涉及一种杂化抗癌药物及其制备方法与应用。
背景技术:
在过去的几年中,为了使药物具有双重或多重作用,将两个药效团杂化在一个药物分子里的杂化药物已经成为新药开发中的新热点。由于癌症、神经变性疾病等许多严重疾病的复杂性,这些疾病很难通过一个高度选择性的药效团来治愈,这也是杂化药物近年受到特别关注的原因。相较于普通药物,杂化药物可以用来改善分子亲和力和选择性,降低副作用等。杂化分子的组成单位可以是人工设计的或者天然有机小分子、多肽、核酸等。针对作用靶点,这些药效团之间有望起着协同的作用。
细胞凋亡是多细胞生物体内由一系列有序信号流调控的细胞程序性死亡行为,是机体消除有害细胞、防止细胞过度增殖的正常生理过程,在生物体的生长发育、维持内环境的稳定中起着重要的作用。研究表明,细胞凋亡机制失调是癌症等疾病的重要病理学特征。在过去的几十年间,尽管癌症治疗取得了一些进展,但肿瘤细胞对各种细胞毒刺激产生耐受,细胞凋亡紊乱和失调,仍然是癌症治疗不得不克服的主要障碍。
凋亡抑制蛋白(Inhibitor of apoptosis,IAP)是细胞凋亡通路内抑制细胞死亡的关键调节因子。凋亡抑制蛋白在多种肿瘤细胞中过度表达。目前,IAPs家族包括八个成员,分别是Cp-IAP、Op-IAP、XIAP、c-IAP1、c-IAP2、NAIP、Livin/ML-IAP和Survivin,其中四种凋亡抑制蛋白XIAP、c-IAP1、c-IAP2以及ML-IAP直接参与调节细胞凋亡,这些IAPs蛋白主要是通过抑制半胱天冬酶(caspase)来抑制细胞凋亡。
Smac/DIABLO(第二个线粒体来源的胱氨酸酶激活剂/低等电点IAP直接结合蛋白)是一种从线粒体中释放至细胞质的IAP内源性拮抗剂。Smac通过其N端的Ala-Val-Pro-Ile(AVPI)四肽基序与IAPs结合,从而移除IAPs对细胞凋亡的抑制作用,促进细胞凋亡。以AVPI四肽分子为先导化合物,设计和合成非肽类小分子Smac模拟物,以IAPs为靶点,开发诱导细胞凋亡的抗癌药物成为癌症治疗领域的研究热点。
技术实现要素:
为克服上述现有技术中存在的缺点与不足,本发明的首要目的在于提供一种杂化抗癌药物。所述的杂化抗癌药物能拮抗细胞凋亡抑制蛋白作用以及产生醌的甲基化物中间体。
本发明的另一目的在于提供上述杂化抗癌药物的制备方法。
本发明的再一目的在于提供上述杂化抗癌药物的应用。
本发明的目的通过下述技术方案实现:一种杂化抗癌药物,其结构式如式Ⅰ所示:
其中:
X选自-SCH2-、-CH=CH-或-(CH2)2-取代基中的一种:
Y选自-O-或-OCH2O-中的一种;
R1选自取代基中的一种;
R2选自-CN或-ONO2中的一种。
上述的杂化抗癌药物的制备方法,包括下述步骤:
1)将化合物A和无机碱溶于极性有机溶剂中,悬浮液搅拌1~30分钟,然后加入碘甲烷或碘乙烷,混合液在惰性气体的保护下,室温搅拌4~12小时,浓缩,所得残留物溶于非极性有机溶剂中,有机相用水洗涤,干燥剂干燥,浓缩所得粗产品过柱纯化,得到无色油状物B;
其中,所述的化合物A、无机碱、碘甲烷或碘乙烷的摩尔比为:1:1~10:1~5;
2)-10~15℃下,将TFA的氯代烃溶液加入至化合物B的氯代烃溶液中,混合液搅拌0.3~5小时,浓缩得到粗产品胺C,-10~15℃下,将化合物C溶于干燥的氯代烃溶液中,加入叔丁氧羰基保护的氨基酸、缩合剂和添加剂,然后加入有机碱调节反应液pH至碱性,混合液室温搅拌过夜,反应液依次用稀酸溶液、稀碱溶液、饱和食盐水洗涤,干燥,减压浓缩所得粗产品过柱纯化,得到无色油状物D;
其中,所述的化合物C、叔丁氧羰基保护的氨基酸、缩合剂、添加剂的摩尔比为1:1~5:1~20:0.25~20;
3)将化合物D溶于极性有机溶剂和水的混合溶剂中,加入强碱,悬浮液室温搅拌2~7小时,浓缩,残留物用水稀释,用稀盐酸溶液调节pH值至酸性,水溶液用非极性有机溶剂萃取三次,合并有机相,干燥剂干燥,浓缩得到无色油状物E;
其中,所述的化合物D、强碱的摩尔比为1:1~5;
4)-10~15℃下,将化合物E溶于干燥氯代烃溶液中,加入有机碱或缩合剂和有机碱的混合氯代烃溶液,然后加入2-(4-(氯代甲氧基)苯基)乙腈或2-(4-(氯代甲氧基)苯基)甲基硝酸酯或2-(4-羟基苯基)乙腈或2-(4-羟基苯基)甲基硝酸酯,混合液在惰性气体保护下,室温搅拌过夜,浓缩得到粗产品F,-10~15℃,将TFA加入至化合物F的氯代烃溶液中,混合液搅拌0.1~5小时,浓缩,高效液相纯化,冷冻干燥,得到白色成品G;
其中,所述的化合物E、2-(4-(氯代甲氧基)苯基)乙腈或2-(4-(氯代甲氧基)苯基)甲基硝酸酯或2-(4-羟基苯基)乙腈或2-(4-羟基苯基)甲基硝酸酯、有机碱、缩合剂的摩尔比为1:1~5:1~20:1~20。
优选的,上述的杂化抗癌药物的制备方法,所述的化合物A的结构通式为:其中,X为-SCH2-、-CH=CH-或-(CH2)2-中的一种。
优选的,上述的杂化抗癌药物的制备方法,所述的无机碱为K2CO3、Na2CO3、KHCO3、NaHCO3、KOH、NaOH或LiOH中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的强碱为KOH、NaOH、Ca(OH)2、NaH、KH或LiOH中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的极性有机溶剂为DMF、DMSO、THF或MeCN中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的非极性有机溶剂为乙酸乙酯、二氯甲烷、氯仿或乙醚中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的惰性气体为氮气或氩气。
优选的,上述的杂化抗癌药物的制备方法,所述的氯代烃溶液为二氯甲烷或氯仿。
优选的,上述的杂化抗癌药物的制备方法,所述的叔丁氧羰基保护的氨基酸为N-叔丁氧羰基-L-丙氨酸、(S)-2-(叔丁氧羰基)氨基丁酸、(S)-2-((叔丁氧羰基)甲基氨基)丙酸或(S)-2-((叔丁氧羰基)甲基氨基)丁酸中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的缩合剂为BOP、DCC、EDCI、DIC、PyBOP、TBTU、HBTU、HATU或TATU中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的添加剂为HOBt、HOOBt或HOSu中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的有机碱为三乙胺、吡啶、DBU、DIEA或N-甲基吗啉中的一种或多种。
优选的,上述的杂化抗癌药物的制备方法,所述的干燥剂为无水硫酸镁、无水硫酸钠、无水碳酸钾或无水氯化钙中的一种。
上述的杂化抗癌药物作为抗癌药物的应用。与现有技术相比,本发明的提供的技术方案与XIAP-BIR3的结合亲和力较强,非肽特征更加明显,且有望作用于多个抗癌靶点。
本发明相对于现有技术具有如下的优点及效果:
本发明为了加强Smac模拟物的抗癌活性,用一个新的基团替代Smac模拟物碳端的疏水性基团,设计和合成了一类杂化抗癌药物。所引入的新基团不仅与凋亡抑制蛋白作用,还能在酯酶的水解作用下,生成醌的甲基化物。醌的甲基化物中间体高度亲电,可以参与多项生理过程,诱导细胞死亡以及抑制细胞生长。因此,该类杂化药物包括两个药效团:一个药效团与IAPs结合,拮抗IAPs对细胞凋亡的抑制作用;另一个药效团产生醌的甲基化物中间体,参与多项生理过程。因此,本发明所设计及合成的杂化抗癌药物与XIAP-BIR3的结合亲和力较强,非肽特征更加明显,可以作用于多个靶点,起着多重功效。
附图说明
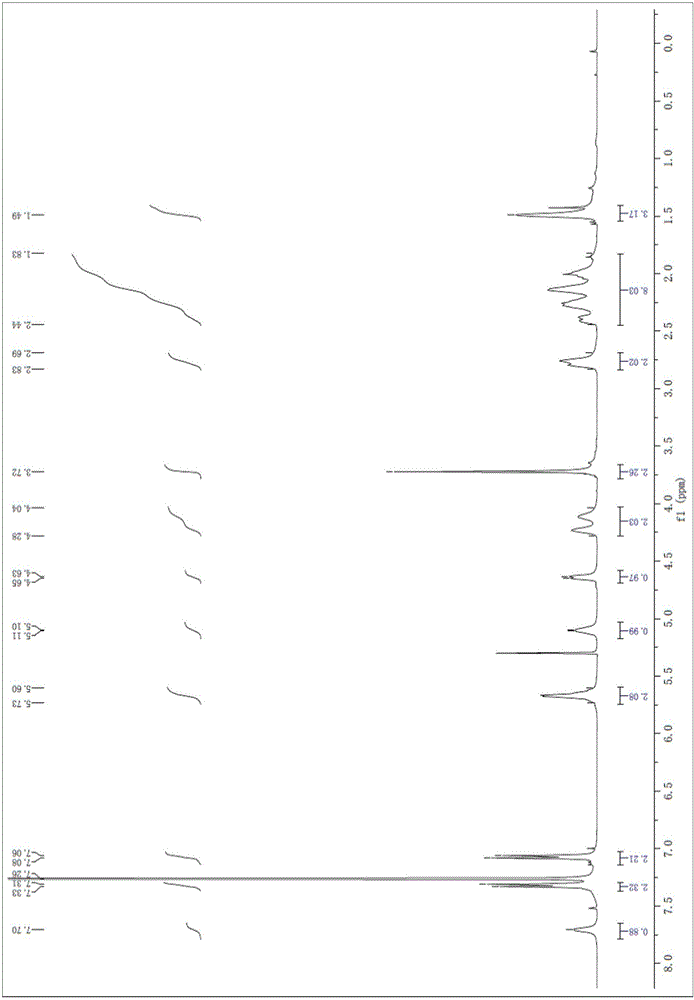
图1是化合物G1的1H NMR检测谱图;
图2是化合物G2的1H NMR检测谱图;
图3是化合物G3的1H NMR检测谱图;
图4是化合物G1与XIAP-BIR3竞争性结合抑制曲线;
图5是化合物G1与TRAIL对MDA-MB-231细胞凋亡的协同作用。
具体实施方式
下面结合具体实施方式,对本发明的权利要求做进一步的详细说明,但是不构成对本发明的任何限制,任何人在本发明权利要求保护范围内所做的有限次的修改,仍在本发明的权利要求保护范围之内。
所有试剂未经特别说明,均为市售分析纯,无进一步纯化,直接使用。无水无氧反应的玻璃仪器在使用前,在烘箱中干燥,反应在Ar保护下进行。溶剂重蒸均在Ar保护下进行,THF在金属钠和二苯甲酮的条件下干燥,二氯甲烷、乙酸乙酯用氢化钙干燥。柱层析使用63~200目硅胶,薄层层析使用Merck TLC silica gel 60F254塑料色谱板,用254nm紫外光照、或碘蒸气、或2%茚三酮的乙醇溶液显色检测。核磁共振谱使用Bruker AVANCE III 400NMR谱仪测定,化学位移值以ppm为单位记录。质谱使用LCT Premier XE型飞行时间质谱仪测定。旋光值用JASCO P-1010旋光仪测定。
高效液相色谱(HPLC)条件:(1)制备型HPLC:Perkin Elmer Series 200UV/VIS检测器;Perkin Elmer Series 200二相泵;真空除气器;Gilson FC203b馏分接收仪;(2)分析型HPLC:Perkin Elmer Series 225自动进样器;Perkin Elmer Series 220UV/VIS检测器;Perkin Elmer Series 200四相泵;真空除气器。流动相:(1)流动相A:水+0.045%TFA;(2)流动相B:10%水+90%乙腈+0.038%TFA。
实施例1(杂化抗癌药物G1)
化合物A1(A1的结构式见式Ⅱ中所示)的合成方法参照中国专利201610036840.2第[0046]至[0083]段的制备方法。
(5R,8S,10aR)-5-((叔丁氧甲酰胺基)-6-羰基硫杂氮杂卓[1,5]并吡咯烷[2,1-d]-8-羧酸甲酯(化合物B1)的制备:
将化合物A1(36mg,0.10mmol)和K2CO3(69mg,0.50mmol)溶于DMF(2mL)中。悬浮液搅拌15分钟,然后加入碘甲烷(31μL,0.50mmol)。混合液在Ar保护下,室温搅拌8小时,浓缩,所得残留物溶于乙酸乙酯中。有机相用水洗涤,无水硫酸镁干燥,浓缩所得粗产品过柱纯化(环己烷/乙酸乙酯=3/1),得到无色油状物B1(28mg,78%),反应化学方程式见式Ⅱ所示。
1H NMR(400MHz,CDCl3):δ5.87(d,J=6.9Hz,1H),4.71-4.66(m,1H),4.61-4.56(m,1H),4.49(t,J=9.0Hz,1H),3.74(s,3H),2.96-2.88(m,2H),2.75-2.67(m,2H),2.43-2.36(m,1H),2.14-1.91(m,3H),1.80(dd,J=11.7,6.7Hz,1H),1.75-1.66(m,1H),1.40(s,9H);13C NMR(100MHz,CDCl3):δ173.29,170.41,154.97,80.06,59.67,56.82,56.34,52.63,38.43,31.40,30.49,28.64,27.04.
(5R,8S,10aR)-5-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-6-羰基硫杂氮杂卓[1,5]并吡咯烷[2,1-d]-8-羧酸甲酯(化合物D1)的制备:
-10℃下,将TFA(300μL)的二氯甲烷溶液(1mL)加入至化合物B1(28mg,0.08mmol)的二氯甲烷溶液(2mL)中。混合液搅拌5h,浓缩得到化合物胺C1。-10℃下,将所得的化合物C1溶于干燥的二氯甲烷(3mL)中,加入Boc-Ala-OH(15mg,0.08mmol)、EDCI(307mg,1.6mmol)、HOBt(216mg,1.6mmol),然后加入DIEA调节反应液至碱性。混合液室温搅拌过夜,反应液依次用稀盐酸溶液、饱和碳酸氢钠溶液、饱和食盐水洗涤。有机相用无水硫酸镁干燥,减压浓缩,所得粗产品过柱纯化(环己烷/丙酮=3/1),得到无色油状物D1(25mg,两步反应总产率73%),反应化学方程式见式Ⅲ。
1H NMR(400MHz,CDCl3):δ7.33(d,J=4.2Hz,1H),5.02(d,J=6.1Hz,1H),4.81-4.76(m,1H),4.72-4.67(m,1H),4.49(t,J=9.0Hz,1H),4.19-4.05(m,1H),3.74(s,3H),2.98-2.86(m,2H),2.76-2.65(m,2H),2.44-2.37(m,1H),2.15-1.92(m,3H),1.81(dd,J=11.8,6.8Hz,1H),1.72(tt,J=13.9,4.5Hz,1H),1.42(s,9H),1.32(d,J=7.1Hz,3H);13C NMR(100MHz,CDCl3):δ173.18,171.85,169.90,155.52,80.38,59.72,56.39,55.76,52.67,50.63,38.01,37.09,31.54,31.38,28.62,27.02,19.13.
(5R,8S,10aR)-5-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-6-羰基硫杂氮杂卓[1,5]并吡咯烷[2,1-d]-8-羧酸(化合物E1)的制备:
将化合物D1(25mg,0.06mmol)溶于MeCN/H2O(1:1,2mL)的混合溶剂中,加入KOH(3.3mg,0.06mmol)。悬浮液室温搅拌2小时,浓缩,残留物用水稀释,用稀盐酸溶液调节pH值至2。水溶液用乙酸乙酯萃取三次,合并有机相,无水硫酸镁干燥,浓缩得到无色油状物E1(23mg,95%)。无需进一步纯化,直接进行下一步反应。反应化学方程式见式Ⅳ。
1H NMR(400MHz,MeOD):δ4.84(dd,J=10.1,2.3Hz,1H),4.76-4.71(m,1H),4.48(t,J=8.8Hz,1H),4.08(q,J=7.2Hz,1H),2.98-2.72(m,4H),2.49-2.43(m,1H),2.20-2.00(m,3H),1.86(dd,J=11.1,6.1Hz,1H),1.80-1.72(m,1H),1.46(s,9H),1.33(d,J=7.2Hz,3H);13C NMR(100MHz,MeOD):δ177.10,175.63,172.01,158.56,81.59,62.07,58.77,57.62,52.57,38.94,38.71,32.69,32.16,29.55,28.76,18.87.
(5R,8S,10aR)-(4-(氰甲基)苯氧基)甲基5-((S)-2-氨基丙酰胺基)-6-羰基硫杂氮杂卓[1,5]并吡咯烷[2,1-d]-8-羧酸酯三氟乙酸盐(G1)的制备:
-10℃下,将化合物E1(23mg,0.06mmol)溶于干燥二氯甲烷溶液(3mL)中,加入三乙胺(8μL,0.06mmol),然后加入2-(4-(氯代甲氧基)苯基)乙腈(11mg,0.06mmol)的干燥二氯甲烷溶液(2mL)。混合液在Ar保护下室温搅拌过夜,浓缩得到粗产品F 1。-10℃下,将化合物F1溶于二氯甲烷(2mL)中,加入TFA(300μL)的二氯甲烷溶液(1mL),混合液搅拌5小时,浓缩所得残留物经HPLC纯化,冷冻干燥,得到白色固体G1(7.8mg,两步总产率23%)。反应化学方程式见式Ⅴ。化合物G1的1H NMR检测谱图如图1所示。
1H NMR(400MHz,D2O):δ7.27(d,J=8.8Hz,2H),7.01(d,J=8.8Hz,2H),5.84,5.76(ABq,J=6.8Hz,2H),4.49-4.44(m,1H),4.35(t,J=9.1Hz,1H),3.97(q,J=7.1Hz,1H),3.76(s,2H),2.76(dd,J=14.1,3.0Hz,1H),2.67(dt,J=15.6,4.5Hz,1H),2.32-2.24(m,3H),2.07-1.97(m,1H),1.93-1.83(m,1H),1.72-1.62(m,3H),1.36(d,J=7.1Hz,3H);13C NMR(100MHz,D2O):δ172.82,172.77,170.92,170.88,169.81,169.77,155.11,155.05,129.72,129.67,125.71,125.63,120.19,120.13,117.22,117.14,86.23,86.15,60.62,60.57,57.94,57.88,54.70,54.65,48.91,48.83,35.94,35.88,34.73,34.66,30.89,30.83,29.48,29.42,26.47,26.41,22.03,21.98,16.44,16.39;MS(ESI):m/z 461.2(M+H)+;HR ESI MS for C22H29N4O5S required 461.1859,found:461.1880.
实施例2(杂化抗癌药物G2)
化合物A2(A2的结构式见式Ⅵ中所示)的合成方法参照Duggan,H.M.等人在Organic&biomolecular chemistry(《有机和生物分子化学》)杂志2005年第3期2287页报道的方法。
(3S,6S,10aR)-6-(叔丁氧甲酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸乙酯(化合物B2)的制备:
将化合物A2(32mg,0.10mmol)和Na2CO3(106mg,1.00mmol)溶于DMSO(2mL)中。悬浮液搅拌30分钟,然后加入碘乙烷(8μL,0.10mmol)。混合液在N2保护下,室温搅拌4小时,浓缩,所得残留物溶于二氯甲烷中。有机相用水洗涤,无水硫酸钠干燥,浓缩所得粗产品过柱纯化(环己烷/乙酸乙酯=6/1),得到无色油状物B2(25mg,72%)。反应化学方程式见式Ⅵ。
1H NMR(400MHz,CDCl3):δ5.83-5.72(m,1H),5.72-5.62(m,1H),5.56(d,J=7.5Hz,1H),4.86-4.76(m,1H),4.50-4.39(m,1H),4.12(q,J=7.1Hz,3H,containing 1H),2.76-2.70(m,2H),2.45-2.33(m,1H),2.32-2.19(m,1H),2.15-2.06(m,1H),2.06-1.98(m,1H),1.98-1.82(m,2H),1.39(s,9H),1.22(t,J=7.1Hz,3H);13C NMR(100MHz,CDCl3):δ171.89,171.05,155.23,129.32,125.92,79.58,60.97,60.38,58.74,51.91,35.37,33.03,32.97,28.42(3C),27.29,14.21.
(3S,6S,10aR)-6-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸乙酯(化合物D2)的制备:
0℃下,将TFA(300μL)的二氯甲烷溶液(1mL)加入至化合物B2(28mg,0.08mmol)的二氯甲烷溶液(2mL)中。混合液搅拌2.5h,浓缩得到化合物胺C2。0℃下,将所得的化合物C2溶于干燥的二氯甲烷(3mL)中,加入Boc-Ala-OH(38mg,0.20mmol)、BOP(35mg,0.08mmol)、HOOBt(3mg,0.02mmol),然后加入NEt3调节反应液至碱性。混合液室温搅拌过夜,反应液依次用饱和氯化铵溶液、饱和碳酸氢钾溶液、饱和食盐水洗涤。有机相用无水硫酸钠干燥,减压浓缩,所得粗产品过柱纯化(环己烷/乙酸乙酯=5/1),得到无色油状物D2(26mg,两步反应总产率77%)。反应化学方程式见式Ⅶ。
1H NMR(400MHz,CDCl3):δ7.06(d,J=5.7Hz,1H),5.93-5.78(m,1H),5.78-5.62(m,1H),5.13-4.90(m,2H),4.46(dd,J=8.9,3.9Hz,1H),4.28-4.05(m,4H),2.87-2.66(m,2H),2.54-2.38(m,1H),2.38-2.24(m,1H),2.24-2.08(m,1H),2.08-1.82(m,3H),1.43(s,9H),1.33(d,J=7.0Hz,3H),1.26(t,J=7.1Hz,3H).
(3S,6S,10aR)-6-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸(化合物E2)的制备:
将化合物D2(25mg,0.06mmol)溶于THF/H2O(1:1,2mL)的混合溶剂中,加入LiOH(4.3mg,0.18mmol)。悬浮液室温搅拌4.5小时,浓缩,残留物用水稀释,用稀盐酸溶液调节PH值至2。水溶液用二氯甲烷萃取三次,合并有机相,无水硫酸钠干燥,浓缩得到无色油状物E2(22mg,93%)。无需进一步纯化,直接进行下一步反应。反应化学方程式见式Ⅷ。
1H NMR(400MHz,CDCl3):δ9.55(bs,1H),7.63(bs,1H),7.50(bs,1H),5.96-5.62(m,2H),5.35-5.14(m,1H),4.71-4.45(m,1H),4.33-4.08(m,2H),3.00-2.70(m,2H),2.54-2.32(m,1H),2.32-2.14(m,2H),2.14-1.98(m,2H),1.98-1.84(m,1H),1.44(d,J=5.0Hz,9H),1.37(dd,J=6.9,3.5Hz,3H);13C NMR(100MHz,CDCl3):δ172.33,155.85,129.25,125.94,80.54,60.73,59.29,50.52,49.93,34.59,33.07,32.78,28.46,26.59,19.56,14.33.
(3S,6S,10aR)-(4-乙氰基苯基)-6-((S)-2-氨基丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烷-3-羧酸酯三氟乙酸盐(G2)的制备:
0℃下,将化合物E2(22mg,0.06mmol)溶于干燥的氯仿(5mL)中,加入EDCI(115mg,0.60mmol)和DIEA(99μL,0.60mmol)的混合氯仿溶液,然后加入2-(4-羟基苯基)乙腈(24mg,0.18mmol),混合液室温搅拌过夜。减压抽干溶剂,得到淡黄色油状物F2。将F2溶于氯仿(3mL),冷至0℃,滴加TFA(0.5mL),2.5h后,蒸干溶剂,浓缩所得残留物经HPLC纯化,冷冻干燥,得到白色固体G2(3.1mg,两步反应总产率10%)。反应化学方程式见式Ⅸ。化合物G2的1H NMR检测谱图如图2所示。
1H NMR(400MHz,CDCl3):δ7.70(bs,1H),7.32(d,J=8.3Hz,2H),7.07(d,J=8.3Hz,2H),5.85-5.51(m,2H),5.16-5.04(m,1H),4.64(d,J=5.9Hz,1H),4.33-4.16(m,1H),4.16-3.99(m,1H),3.72(s,2H),2.86-2.59(d,J=14.8Hz,2H),2.51-2.34(m,1H),2.25-2.20(m,2H),2.20-2.07(m,3H),2.06-1.90(m,2H),1.55-1.43(m,3H).MS(ESI):m/z 411.2(M+H)+;HR ESI MS for C22H27N4O4required 411.1954,found:411.1932.
实施例3(杂化抗癌药物G3)
化合物A2(A2的结构式见式Ⅹ中所示)的合成方法参照Duggan,H.M.等人在Organic&biomolecular chemistry(《有机和生物分子化学》)杂志2005年第3期2287页报道的方法。
(3S,6S,10aR)-6-(叔丁氧甲酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸乙酯(化合物B2)的制备:
将化合物A2(32mg,0.10mmol)和KOH(6mg,0.10mmol)溶于THF(2mL)中。悬浮液搅拌1分钟,然后加入碘乙烷(20μL,0.25mmol)。混合液在N2保护下,室温搅拌12小时,浓缩,所得残留物溶于氯仿中。有机相用水洗涤,无水碳酸钾干燥,浓缩所得粗产品过柱纯化(环己烷/乙酸乙酯=6/1),得到无色油状物B2(29mg,82%)。反应化学方程式见式Ⅹ。
1H NMR(400MHz,CDCl3):δ5.83-5.72(m,1H),5.72-5.62(m,1H),5.56(d,J=7.5Hz,1H),4.86-4.76(m,1H),4.50-4.39(m,1H),4.12(q,J=7.1Hz,3H,containing 1H),2.76-2.70(m,2H),2.45-2.33(m,1H),2.32-2.19(m,1H),2.15-2.06(m,1H),2.06-1.98(m,1H),1.98-1.82(m,2H),1.39(s,9H),1.22(t,J=7.1Hz,3H);13C NMR(100MHz,CDCl3):δ171.89,171.05,155.23,129.32,125.92,79.58,60.97,60.38,58.74,51.91,35.37,33.03,32.97,28.42(3C),27.29,14.21.
(3S,6S,10aR)-6-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸乙酯(化合物D2)的制备:
15℃下,将TFA(300μL)的氯仿溶液(1mL)加入至化合物B2(28mg,0.08mmol)的氯仿溶液(2mL)中。混合液搅拌0.3h,浓缩得到化合物胺C2。15℃下,将所得的化合物C2溶于干燥的氯仿(3mL)中,加入Boc-Ala-OH(76mg,0.40mmol)、DCC(165mg,0.80mmol)、HOSu(92mg,0.80mmol),然后加入DBU调节反应液pH至碱性。混合液室温搅拌过夜,反应液依次用饱和氯化铵溶液、饱和碳酸氢钾溶液、饱和食盐水洗涤。有机相用无水硫酸钠干燥,减压浓缩,所得粗产品过柱纯化(环己烷/乙酸乙酯=5/1),得到无色油状物D2(23mg,两步反应总产率68%)。反应化学方程式见式Ⅺ。
1H NMR(400MHz,CDCl3):δ7.06(d,J=5.7Hz,1H),5.93-5.78(m,1H),5.78-5.62(m,1H),5.13-4.90(m,2H),4.46(dd,J=8.9,3.9Hz,1H),4.28-4.05(m,4H),2.87-2.66(m,2H),2.54-2.38(m,1H),2.38-2.24(m,1H),2.24-2.08(m,1H),2.08-1.82(m,3H),1.43(s,9H),1.33(d,J=7.0Hz,3H),1.26(t,J=7.1Hz,3H).
(3S,6S,10aR)-6-((S)-2-(叔丁氧甲酰胺基)丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烯-3-羧酸(化合物E2)的制备:
将化合物D2(25mg,0.06mmol)溶于MeCN/H2O(1:1,2mL)的混合溶剂中,加入NaOH(12mg,0.30mmol)。悬浮液室温搅拌7小时,浓缩,残留物用水稀释,用稀盐酸溶液调节pH值至2。水溶液用氯仿萃取三次,合并有机相,无水碳酸钾干燥,浓缩得到无色油状物E2(20mg,84%)。无需进一步纯化,直接进行下一步反应。反应化学方程式见式Ⅻ。
1H NMR(400MHz,CDCl3):δ9.55(bs,1H),7.63(bs,1H),7.50(bs,1H),5.96-5.62(m,2H),5.35-5.14(m,1H),4.71-4.45(m,1H),4.33-4.08(m,2H),3.00-2.70(m,2H),2.54-2.32(m,1H),2.32-2.14(m,2H),2.14-1.98(m,2H),1.98-1.84(m,1H),1.44(d,J=5.0Hz,9H),1.37(dd,J=6.9,3.5Hz,3H);13C NMR(100MHz,CDCl3):δ172.33,155.85,129.25,125.94,80.54,60.73,59.29,50.52,49.93,34.59,33.07,32.78,28.46,26.59,19.56,14.33.
(3S,6S,10aR)-(4-硝酸酯亚甲基苯基)-6-((S)-2-氨基丙酰胺基)-5-羰基-1,2,3,5,6,7,10,10a-八氢吡咯并[1,2-a]氮杂环辛烷-3-羧酸酯三氟乙酸盐(G3)的制备:
15℃下,将化合物E2(20mg,0.05mmol)溶于干燥的氯仿(5mL),加入DCC(206mg,1mmol)和DIEA(165μL,1mmol)的混合氯仿溶液,然后加入2-(4-羟基苯基)甲基硝酸酯(42mg,0.25mmol),混合液室温搅拌过夜。减压抽干溶剂,得到淡黄色油状物F3。将F3溶于氯仿(3mL),在15℃下,滴加TFA(0.5mL),0.3h后,蒸干溶剂,浓缩所得残留物经HPLC纯化,冷冻干燥,得到白色固体G3(2.3mg,8%)。反应化学方程式见式ⅩⅢ。化合物G3的1H NMR检测谱图如图3所示。
1H NMR(400MHz,CDCl3):δ7.46-7.29(m,2H),7.27-7.10(m,2H),5.95-5.66(m,2H),5.04-4.80(m,1H),4.67(dd,J=7.7,6.1Hz,1H),4.24-4.10(m,2H),4.07-3.99(m,1H),3.71-3.51(m,1H),2.77-2.47(m,2H),2.47-2.23(m,1H),2.20-1.45(m,7H),1.54(d,J=6.4Hz,3H).MS(ESI):m/z 447.2(M+H)+;HR ESI MS for C21H27N4O7required 446.1801,found:446.1859.
化合物G4~G48可采用与制备G1或G2相同的方法制得,所不同是:
采用BOP、或DCC、或PyBOP、或DIC、或TBTU、或HBTU、或HATU、或TATU替换实施例1中的EDCI;
采用HOOBt、或HOSu替换实施例1中的HOBt;
采用2-(4-(氯代甲氧基)苯基)甲基硝酸酯或2-(4-羟基苯基)乙腈或2-(4-羟基苯基)甲基硝酸酯替换实施例1中的2-(4-(氯代甲氧基)苯基)乙腈;
采用(S)-2-(叔丁氧羰基)氨基丁酸、或(S)-2-((叔丁氧羰基)甲基氨基)丙酸、或(S)-2-((叔丁氧羰基)甲基氨基)丁酸替换实施例1中的N-叔丁氧羰基-L-丙氨酸。
采用吡啶、或DBU、或N-甲基吗啉替换实施例1中的DIEA和三乙胺。
为了更好的说明本发明的效果,下面结合给出本发明的实验证明例:
1、实验证明例1
X-连锁凋亡抑制蛋白(XIAP)是IAP蛋白家族的重要成员,XIAP通过直接与caspase-3、caspase-7、caspase-9结合,来抑制caspase的活性,从而抑制细胞凋亡。本发明以G1、G2以及G3为例,在荧光偏振试验中,分别测定G1、G2以及G3拮抗X-连锁凋亡抑制蛋白(XIAP)的BIR3域活性的能力。
1.1实验材料及仪器
(1)荧光标记的多肽配体
由同事合成七肽分子H-AVPFAQK-(6-FAM)-NH2,在其赖氨酸支链的ε氨基上做荧光标记。该荧光肽的结构由ESI-MS确认,经HPLC分析,其纯度高于98%。荧光肽溶于超纯水中,其溶液浓度经紫外吸收法确定为467μM。溶液每10μL分装,用锡纸包裹避光,-20℃保存。
(2)XIAP-BIR3重组蛋白
XIAP-BIR3重组蛋白购于R&D Systems Inc.。蛋白是大肠杆菌来源的,含Asn252-Thr356残基,带一个N端Met和6-His标签。每50μg分装,-20℃下保存在50μL的pH值为8.0,含50mM Tris–HCl、300mM KCl、50μM醋酸锌、1mM DTT的缓冲溶液中。
(3)反应缓冲液
本测试使用的反应缓冲液包含20mM Hepes、2mM dithiothreitol(DTT)、100mM NaCl、0.1%Bovine serum albumin(BSA),pH仪调节其pH值至7.4。所用试剂均购于Sigma-Aldrich公司,纯度为最高级别。
(4)样品溶液的配制
首先将化合物G1、G2以及G3分别溶于超纯水,配制为10mM的母液,然后将母液稀释至所需浓度。
(5)实验仪器
Perkin Elmer EnVision多标记微孔板检测仪(EnVision Multilabel Reader),pH计,紫外吸收检测仪,384孔检测板。
1.2实验方法
(1)准备384孔检测板
首先,将2.5nM的荧光肽和250nM的XIAP-BIR3蛋白加入至反应缓冲液中,形成测试缓冲溶液,并孵化半小时。与此同时,将样品母液稀释成一系列浓度,浓度范围为5mM至5nM。
然后,在384孔检测板上,每一个浓度点三个重复样。首先,用移液枪向384孔检测板的孔中加入16μL的测试缓冲溶液,再加入4μL样品溶液。混合之后,孔中溶液体积变为20μL,于是,荧光肽和XIAP-BIR3的浓度分别变为最佳测试浓度,即2nM和200nM。样品的最终浓度变为1mM至1nM。将384孔检测板室温孵化20~30分钟,用微孔板检测仪读数。然后将检测板再孵化20~30分钟,再次读数。
(2)数据处理
所有数据均运用GraphPad Prism的非线性回归曲线拟合程序分析。
1.3结果与讨论
荧光肽和XIAP-BIR3蛋白的最终测试浓度分别固定为2nM和200nM,所测试样品的最终浓度范围为1mM至1nM。化合物G1与XIAP-BIR3竞争性结合抑制曲线如图4所示,以mP值为Y轴,log C为X轴(C为测试样品的浓度)。结果表明,化合物G1具有较好的与XIAP-BIR3的结合亲和力。由于化合物G1对XIAP-BIR3竞争性结合抑制,其结合曲线为S型,即mP值随测试样品浓度的增加而减小,当样品浓度足够低时,mP值达到最大值,不再变化。反之,当样品浓度足够高时,mP值达到最小值,抑制饱和。所测数据经GraphPad Prism分析,得出化合物G1的IC50值为3.6μM。
在同样的条件下,用同样的检测方法,测得G2的IC50值为6.7μM,G,3的IC50值为8.3μM。此外,阳性对照物——四肽分子AVPI(Ala-Val-Pro-Ile)的IC50值为0.8μM。虽然G1、G2以及G3的IC50值不如AVPI,但是化合物G1、G2以及G3的非肽特征更加明显,且有望作用于多个抗癌靶点。荧光偏振试验结果表明,化合物G1、G2以及G3是一种新型、较高效的XIAP非肽拮抗剂。
2、实验证明例2
以G1为例,在MDA-MB-231乳腺癌细胞株中,测定G1与另一种抗癌药物TRAIL一起使用,对细胞凋亡的协同作用。
2.1实验材料及方法
(1)细胞培养及试剂
所有化合物溶于DMSO中,配制为浓度为10mM的储备溶液。细胞在补充10%胎牛血清(FBS)、青霉素(100U/mL)、链霉素(100mg/mL)的DMEM培养基中培养。TRAIL购于PeproTech Inc.。
(2)3-(4,5-二甲基噻唑-2)-2,5-二苯基四氮唑溴盐(MTT)试验
细胞存活率用MTT法测试。首先,将细胞置于96孔板(1×106细胞/孔),然后加入相应浓度的所测化合物和TRAIL,在培养皿中维持24、48、96小时。最后,加入20μL的MTT(5mg/mL的磷酸盐缓冲液)加入至细胞培养皿中,37℃下孵化2小时。所得晶体溶于200μL的DMSO中,用Vector3在490nm波长下测量吸收强度。
2.2结果与讨论
首先,在MDA-MB-231细胞株中,运用MTT试验,测试了10ng/mL、50ng/mL、100ng/mL三个浓度的TRAIL单独使用时对细胞凋亡的影响。结果如图5所示,随着时间的增长,以及随着TRAIL浓度的增大,细胞存活率降低。但是,总的来说,即使在100ng/mL的浓度条件下,TRAIL对MDA-MB-231癌细胞的细胞凋亡作用也不是太明显,细胞培养96小时后,仍有65%的癌细胞存活。
然后,运用MTT试验,测试了5μM G1分别与10ng/mL、50ng/mL、100ng/mL三个浓度的TRAIL共同作用,对细胞凋亡的影响。结果如图5所示,细胞培养48小时后,化合物G1与TRAIL显示出良好的剂量依存性的协同作用,而这种协同作用在较高浓度下显得更为明显。根据所得结果,可以推断出细胞凋亡越活跃,这种协同作用越明显。譬如,96小时孵化后,100ng/mL的TRAIL单独使用,65%细胞存活,然而,TRAIL与5μM G1一起使用时,仅31%的细胞存活,抗癌活性大大提高。
试验结果表明,该类杂化药物可以有效地拮抗IAPs的作用,在MDA-MB-231乳腺癌细胞株中,可以使原本对TRAIL产生药耐受的MDA-MB-231细胞变得对TRAIL敏感,从而加速细胞凋亡。该类杂化药物可以作为新型抗癌药物的先导化合物,用于新型抗癌药物的开发和研究中。
缩略词表(按字母顺序)
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!
- 1‑(2‑吡啶)‑9‑(2‑苯基乙基)‑β‑咔啉的硝酸铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(3‑苯基丙基)‑β‑咔啉的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(4‑甲基苄基)‑β‑咔啉的硝酸铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑丁基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑己基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑辛基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑壬基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 以1‑(2‑吡啶)‑9‑异戊基‑β‑咔啉为配体的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(萘‑2‑甲基)‑β‑咔啉的氯化铜配合物及其合成方法和应用与制造工艺
- 1‑(2‑吡啶)‑9‑(2‑乙氧基乙基)‑β‑咔啉的氯化铜配合物及合成方法和应用与制造工艺