一种O‑取代羟胺荧光衍生化试剂的制备与纯化方法与流程
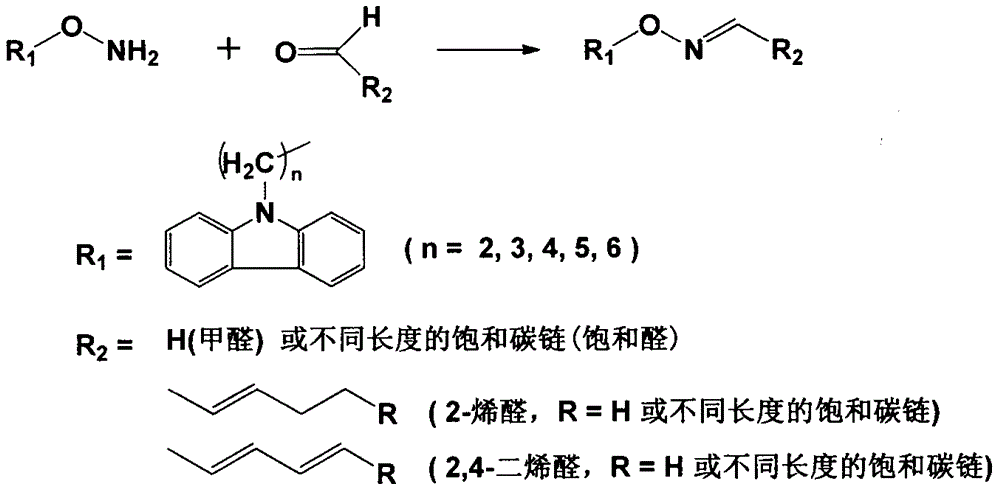
本发明涉及一种O-取代羟胺荧光衍生化试剂的制备与纯化方法,纯化后的试剂可用于醛类化合物或含醛基的物质的标记和衍生化分析测定。
背景技术:
:近年来醛类化合物日渐引起人们的关注,例如室内空气中的甲醛是主要污染物之一,长期吸入易导致多种疾病。甲醛、乙醛、丙醛等小分子醛类化合物也可以作为鉴别地沟油的指标之一,这些醛类物质被吸收进入体内会导致蛋白质、DNA结构发生变化,从而导致各种疾病。体内物质经过代谢也会产生醛类化合物,例如糖类化合物的代谢会生成乙二醛。人体内的醛类化合物主要来自多不饱和脂肪酸的过氧化。多不饱和脂肪酸,例如亚油酸、亚麻酸、花生四烯酸等,广泛存在于人体中,是构成细胞膜和脂蛋白的重要组成成分。因其分子中含有不饱和双键,在酶、过渡金属离子催化下易发生脂质过氧化反应,从而产生一系列活性醛类化合物,例如HNE、ONE、丙烯醛、丙二醛等。这些醛类化合物分子量小、流动性大,传播并扩大了氧化应激相关的组织损伤和功能紊乱。许多种疾病,包括糖尿病、高血压、动脉粥样硬化、阿兹海默症及类风湿性关节炎等都被认为与多不饱和脂肪酸的脂质过氧化有关。现有研究表明醛类化合物在体内可能有双向调节的作用,即少量的醛可能引发机体自身免疫力以提高抗病能力,而过量的醛则会导致多种疾病,如癌症、动脉粥样硬化等。为了研究醛类化合物在体内的作用及其机制,必需准确测定其在体液或组织中的浓度。然而醛基本身反应活性高,不具有紫外吸收或产生荧光的基团,并且其在体内的浓度极低,一般的检测方法无法准确的进行测定。目前常用的醛类化合物的分析方法为柱前衍生化-高效液相色谱法。根据衍生化试剂的官能团不同又分为紫外检测法和荧光检测法,前者如2,4-二硝基苯肼,后者如DNSH、BODIPY等。虽然目前有大量的衍生化试剂可供选择,但其中一些缺乏选择性、灵敏度较低、衍生化产物稳定性差,还有一些会存在羰基化合物的干扰,尤其是生物介质中。含肼基或羟胺取代基的荧光衍生化试剂可提高衍生化反应的选择性和检测灵敏度,能够准确分析体液或组织中的微量醛类化合物,其合成具有实际的科研应用意义。技术实现要素:本发明针对目前醛类化合物衍生化试剂缺乏选择性、灵敏度较低、衍生化产物稳定性差,生物介质中存在羰基化合物的干扰等缺陷,提供一种可用作醛类化合物分析检测的羟胺取代基荧光衍生化试剂的制备和纯化方法。为实现上述目的,本发明的技术方案以咔唑为母体结合下式所述的反应路线,合成目标产物:本发明的制备方法及其特征在于包括有以下步骤:a)中间体I的合成将咔唑溶于反应溶剂中,先加入α,ω-二卤代烷烃(C2-C6),再加入相转移催化剂四正丁基溴化胺,搅拌均匀后缓慢滴入50%的KOH水溶液,滴加完毕于30~90℃下搅拌反应2~6h。反应完成后,减压除去未反应的溶剂得固体,用水洗后过滤,所得产物烘干;其中的反应溶剂可以为α,ω-二卤代烷烃(C2-C6),也可以用四氢呋喃、2-丁酮、二氧六环或甲苯中的任意一种;其中α,ω-二卤代烷的量为咔唑物质的量的3-20倍;四正丁基溴化胺的量为咔唑物质的量的0.01-0.2倍;50%的KOH水溶液为质量浓度,用量为咔唑质量的2-10倍;b)中间体II的合成将步骤a)所得产物用二甲基甲酰胺溶解后加入N-羟基-5-降冰片烯-2,3-二酰亚胺和催化剂碳酸钾,60-75℃下反应2-10h;反应结束后,萃取,无水硫酸钠干燥,减压除去溶剂,经柱层析纯化得白色固体即为中间体II的纯品;其中N-羟基-5-降冰片烯-2,3-二酰亚胺和碳酸钾的量都为咔唑物质的量的1-3倍;c)目标产物的合成将步骤b)得到的中间体II的纯品用乙醇溶解之后与水合肼加热回流反应0.5-2小时,减压除去反应液中未反应的肼和溶剂乙醇,即得目标产物粗品;d)目标产物的纯化向步骤c)目标产物粗品中加少量水和氯仿,加盐酸使水相呈酸性,会有白色蓬松固体析出,过滤可得目标产物的盐酸盐;取盐酸盐溶于水,加氨水使溶液呈碱性,使目标产物游离成为羟胺的形式,用氯仿萃取,氯仿层用无水硫酸钠干燥,过滤后减压除净氯仿即得目标产物O-(ω-(9H-咔唑-9-基)烷基)羟胺的纯品。本发明中衍生化试剂的制备条件反应温度要求低,反应迅速,副产物少,操作简便,纯化方法简单易行,经上述步骤制备的目标产物可用于醛类化合物的衍生化反应实验。本发明制备的荧光衍生化试剂荧光强度大,反应活性高,衍生化速度快,产物稳定,能提高检测灵敏度,是理想的体内醛类化合物分析测定的衍生化试剂。附图说明图1O-取代羟胺衍生化试剂与醛基的反应。图2O-(2-(9H-咔唑-9-基)乙基)羟胺作为衍生化试剂的色谱图。图中衍生化产物为双峰,是由于生成了C=N双键而产生顺反异构体。图3O-(2-(9H-咔唑-9-基)丙基)羟胺作为衍生化试剂的色谱图。C2为乙醛、C3为丙醛、C4为丁醛、C5为戊醛、C6为己醛、C7为庚醛、C8为辛醛、C9为壬醛、C10为癸醛,使用该试剂时每种醛的衍生化产物均显示为单峰,可能是异构体极性一致未能分开。图4O-(2-(9H-咔唑-9-基)丙基)羟胺作为衍生化试剂的色谱图。使用该试剂衍生化不饱和醛时,同样观察到有异构体出现,且分离较好,可能是C=C双键存在的原因。具体实施方式具体实施例1.下面以1,2-二氯乙烷(同时作为反应溶剂)为例说明本发明的内容:a)向装置在磁力搅拌器上的圆底烧瓶中加入5g咔唑,0.96g的四丁基溴化胺和50mL的1,2-二氯乙烷,边搅拌边用滴液漏斗逐滴加入60g50%的KOH水溶液,之后升温至90℃反应6个小时;反应结束后旋蒸除掉未反应的1,2-二氯乙烷得棕色固体,水洗固体后过滤,烘干得浅棕色粉末状固体,为未纯化处理的中间体1。b)将上一步得到的中间体I、5.35gN-羟基-5-降冰片烯-2,3-二酰亚胺、4.14g碳酸钾加入圆底烧瓶中,再加入适量的N,N-二甲基甲酰胺,以没过固体为宜,在磁力搅拌器中75℃下反应10h;反应结束后加水和乙酸乙酯萃取2次,合并乙酸乙酯层,无水硫酸钠干燥,旋转蒸发仪将乙酸乙酯和溶剂N,N-二甲基甲酰胺蒸出,得淡黄色固体,为未纯化处理的中间体II粗品。将中间体II粗品用100-200目的硅胶柱进行洗脱纯化。杂质洗脱用的溶剂为体积比石油醚∶乙酸乙酯=20∶1(v/v),直至将除中间体II的其他物质都洗脱干净后换用乙酸乙酯冲洗即得中间体II的乙酸乙酯溶液,旋蒸除乙酸乙酯后得淡黄色固体约8.9g,即为中间体II纯品。c)将8.9g的中间体II用乙醇溶解,加5mL水合肼,100℃回流反应2h,反应液旋蒸除去乙醇和未反应的水合肼,得淡黄色的液体,即为目标产物粗品。d)向目标产物粗品中加少量水和氯仿,加盐酸至水相呈酸性,有白色蓬松固体析出,过滤除去能溶于有机相的杂质,可得目标产物的盐酸盐;取盐酸盐固体溶于水,加氨水调至碱性使目标产物重新变为羟胺的形式,用氯仿萃取两次,合并氯仿层,无水硫酸钠干燥,减压除净氯仿即得目标产物O-(2-(9H-咔唑-9-基)乙基)羟胺,该化合物物在常温下为液体,放置于-20℃冷冻即得淡黄色固体。具体实施例2.以1,4-二氯丁烷为例,以四氢呋喃为溶剂,说明本发明的内容:a)向装置在磁力搅拌器上的圆底烧瓶中加入5g咔唑,0.96g的四丁基溴化胺,10mL1,4-二氯丁烷,30mL四氢呋喃作为反应溶剂,边搅拌边用滴液漏斗逐滴加入60g50%的KOH水溶液,滴加完毕后升温至50℃反应2个小时;反应结束加入水和乙酸乙酯,摇匀,静置分层,分取乙酸乙酯层用无水硫酸钠干燥,旋蒸除净溶剂得固体,烘干得粉末状固体,为未纯化处理的中间体I。b)将上一步得到的中间体I、5.35gN-羟基-5-降冰片烯-2,3-二酰亚胺、4.14g碳酸钾加入圆底烧瓶中,再加入适量的N,N-二甲基甲酰胺,以没过固体为宜,在磁力搅拌器中60℃下反应6h;反应结束后加水和氯仿摇匀,静置分层,取氯仿层用无水硫酸钠干燥,用旋转蒸发仪将氯仿和溶剂N,N-二甲基甲酰胺蒸出,得灰白色固体,为未纯化处理的中间体II粗品。将中间体II粗品用100-200目的硅胶柱进行洗脱纯化。杂质洗脱用的溶剂为体积比石油醚∶乙酸乙酯=20∶1(v/v),直至将除中间体II的其他物质都洗脱干净后换用乙酸乙酯冲洗即得中间体II的乙酸乙酯溶液,旋蒸除乙酸乙酯后得淡黄色固体约9.8g,即为中间体II纯品。c)将9.8g的中间体II纯品用乙醇溶解,加5mL水合肼,100℃回流反应1.5h,反应液旋蒸除去乙醇和未反应的水合肼,得淡黄色的液体,即为目标产物III粗品。d)向目标产物III粗品中加少量水和氯仿,加盐酸至水相呈酸性,会有白色蓬松固体析出,过滤除去能溶于有机相的杂质,可得目标产物III的盐酸盐;取此盐酸盐固体溶于水,加氨水使目标产物III重新变为羟胺的形式,用氯仿萃取2次,合并氯仿层,无水硫酸钠干燥,减压除净氯仿即得目标产物O-(4-(9H-咔唑-9-基)丁基)羟胺纯品,此产物在常温下为液体,放置于-20℃冷冻即得淡黄色固体。醛的衍生化操作条件及效果1、衍生化试剂的配制:a)准确称取22.6mgO-(2-(9H-咔唑-9-基)乙基)羟胺于10ml容量瓶中,用甲醇稀释至刻度,浓度为10mmol/L;b)准确称取24.0mgO-(2-(9H-咔唑-9-基)丙基)羟胺于10ml容量瓶中,用甲醇稀释至刻度,浓度为10mmol/L;2、醛标准溶液的配制:a)正己醛溶液的配制:取2.46μl的正己醛于10ml容量瓶中,用甲醇稀释至刻度,浓度为2mmol/L;b)饱和混合醛溶液的配制:取11.3μl乙醛,14.3μl正丙醛,17.8μl正丁醛,21.0μl正戊醛,24.6μl正己醛,27.9μl正庚醛,31.3μl辛醛,34.3μl壬醛,37.7μl癸醛于10ml容量瓶中,用甲醇稀释至刻度,配制为浓度20mmol/L的储备液,使用时稀释50倍为浓度0.4mmol/L;c)不饱和混和醛溶液的配制:取11.5μl己烯醛,22.0μl己二烯醛,34.9μl癸二烯醛于10ml容量瓶中,用甲醇稀释至刻度,为储备液,使用时稀释10倍,配制成浓度分别为1mmol/L、2mmol/L、2mmol/L的溶液;3、衍生化操作过程及色谱分离条件:a)O-(2-(9H-咔唑-9-基)乙基)羟胺与正己醛的反应:取10μl2mmol/L正己醛溶液和10μl10mmol/LO-(2-(9H-咔唑-9-基)乙基)羟胺溶液,加入100μl10-4mol/L的磷酸溶液后加水至1000μl,40℃震荡反应15min后,取5μl进样。色谱分离条件:色谱柱为AgilentEclipse-C18键合硅胶柱(4.6mm×75mm,3.5μm),柱温30℃;流动相为乙腈-水(80∶10),流速0.4mL/min;衍生化试剂的荧光激发和发射波长是λex/λem=292/348nm;色谱图见图2。b)O-(2-(9H-咔唑-9-基)丙基)羟胺与饱和混合醛的反应:取10μl0.4mmol/L饱和混合醛溶液和20μl10mmol/LO-(2-(9H-咔唑-9-基)丙基)羟胺溶液,加入100μl10-4mol/L的磷酸溶液后加水至1000μl,40℃震荡反应15min后,取10μl进样。色谱分离条件:色谱柱为AgilentEclipse-C18键合硅胶柱(4.6mm×75mm,3.5μm),柱温30℃;流动相为乙腈-水梯度洗脱,梯度见下表,流速0.4mL/min;衍生化试剂的荧光激发和发射波长是λex/λem=292/348nm;色谱图见图3。时间(min)流动相比例流速(ml/min)070%乙腈0.41095%乙腈0.42595%乙腈0.4c)O-(2-(9H-咔唑-9-基)丙基)羟胺与不饱和混合醛的反应:取30μl不饱和混合醛溶液、2μl2mmol/L正己醛溶液、90μl10mmol/LO-(2-(9H-咔唑-9-基)丙基)羟胺溶液,加入100μl10-4mol/L的磷酸溶液后加水至1000μl,30℃震荡反应2h后,取10μl进样。色谱分离条件:色谱柱为AgilentEclipse-C18键合硅胶柱(4.6mm×75mm,3.5μm),柱温30℃;流动相为乙腈-水梯度洗脱,梯度见下表,流速0.4mL/min;衍生化试剂的荧光激发和发射波长是λex/λem=292/348nm;色谱图见图4。时间(min)流动相比例流速(ml/min)070%乙腈0.41595%乙腈0.42595%乙腈0.4当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1