热稳定性提高的酸性淀粉酶突变体及其制备方法和应用与流程
本发明属于基因工程和酶工程领域,更具体地,本发明涉及热稳定性提高的酸性淀粉酶突变体及其制备方法和应用。
背景技术:
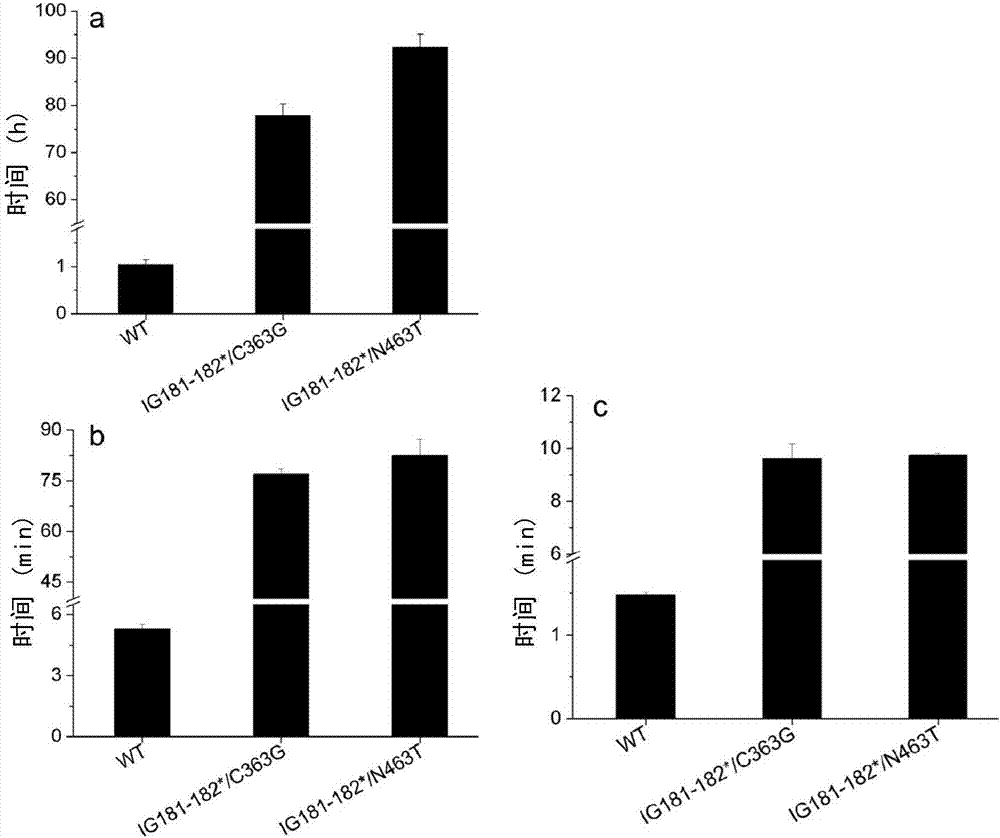
:淀粉是葡萄糖分子通过糖苷键连接而成的高聚物,分为两种类型,直链淀粉和支链淀粉。两种淀粉具有不同的结构和特性,直链淀粉是由多达6000个葡萄糖分子通过α-1,4-糖苷键连接,支链淀粉主链是由较短的含10~60葡萄糖单元通过α-1,4-糖苷键连接,侧链是通过β-1,6-糖苷键与主链连接,侧链长度一般为15-45个葡萄糖单元,通过α-1,4-糖苷键连接起来。从催化专一性上看,把α-淀粉酶归为糖苷水解酶类(glycosylhydrases)的第13家族,属于α-葡萄糖苷水解酶(α-glycosylhydrases,ec3.2.1),α-淀粉酶(α-amylase)又称为液化淀粉酶,其系统名称为α-1,4-葡聚糖-4-葡聚糖水解酶(α-1,4-glucan-glucanhydrolaseec3.2.1.1),常用名α-淀粉酶,又名α-1,4糊精酶,是一种内切酶,能水解淀粉分子中的α-1,4糖苷键,将其任意切成长短不一的短链糊精和少量的低相对分子质量糖类,直链淀粉和支链淀粉均以无规则的形式进行分解,从而使淀粉糊的粘度迅速下降,即“液化”起作用,故α-淀粉酶又称为液化酶。淀粉酶是一种十分重要的酶制剂,大量应用于淀粉转化、洗涤行业、燃料乙醇生产、食品工业、发酵、造纸、纺织品工业和医药行业等,占了整个酶制剂市场份额的30%。α-淀粉酶根据最适反应ph值的不同,分为酸性淀粉酶、中性淀粉酶和碱性淀粉酶。酸性α-淀粉酶为来源于细菌、真菌的淀粉酶,其最适反应ph范围一般为酸性或中性,已广泛应用于食品、化妆品和医药等领域。酸性α-淀粉酶在淀粉加工行业是应用最广泛的,在淀粉液化过程中发挥重要作用。工业上双酶法制糖已经取代了传统的酸法制糖,第一步用到的液化酶即α-淀粉酶至少需要耐受淀粉的糊化温度50-80℃,目前耐受高温的淀粉酶均来自细菌发酵生产,在中国广泛应用的淀粉酶主要有三种,即7658淀粉酶(baa)、中温淀粉酶(bsa)和耐高温α-淀粉酶(bla),均来源于芽孢杆菌,这些酶的特性如表1。表1、三种工业用α-淀粉酶来源于嗜热脂肪芽孢杆菌(b.stearothermophilus)的α-淀粉酶(bsta)也是最早发现的具有良好性能的淀粉酶,在国外也被应用于淀粉加工,例如genencor公司在我国销售的耐高温淀粉酶prime就是用未经基因改造的嗜热脂肪芽孢杆菌生产的。目前制糖工艺广泛采用喷射液化,喷射温度一般在105~108℃,在中间高温维持罐停留5-6min,bla能很好地适应该工艺要求。bla是中国产量最大的淀粉酶,国外最大的酶制剂制造商novozyme和genecor销量最大的α-淀粉酶和spezymepredtm、gc262sptm为其同类产品。市场上应用的α-淀粉酶有一些缺陷,一般α-淀粉酶在ph6.0以下失活,而淀粉浆的自然ph值为4.5,所以应用中需要加氢氧化钠调节ph至6.0进行催化反应,而且由于α-淀粉酶通常是ca2+依赖的,所以需要添加ca2+。因此制糖工艺不仅需要耐受高温的α-淀粉酶,还需要耐酸、ca2+非依赖的α-淀粉酶。鉴于目前耐高温的淀粉酶的需求量大,市售淀粉酶有一些缺陷,开发热稳定性好、耐酸的淀粉酶成了本领域的当务之急。技术实现要素:本发明的目的在于提供热稳定性提高的酸性淀粉酶突变体及其制备方法和应用。在本发明的第一方面,提供具有热稳定性的淀粉酶突变体,所述的淀粉酶突变体基于深海嗜热菌来源的淀粉酶成熟肽,第181位和第182位氨基酸缺失;并且,第463位突变为甘氨酸,或第363位突变为苏氨酸。在一个优选例中,所述的来源于深海嗜热菌来源的淀粉酶成熟肽的氨基酸序列如seqidno:3所示。在本发明的另一方面,提供分离的多核苷酸,所述的核苷酸编码所述的淀粉酶突变体。在本发明的另一方面,提供表达载体,其含有所述的多核苷酸。在本发明的另一方面,提供遗传工程化的宿主细胞,它含有所述的载体,或其基因组中整合有所述的多核苷酸。在本发明的另一方面,提供所述的多肽的制备方法,该方法包含:(i)培养所述的宿主细胞,使之表达所述的淀粉酶突变体;(ii)收集含有所述的淀粉酶突变体的培养物;和(iii)从培养物中分离出所述的淀粉酶突变体。在本发明的另一方面,提供所述的淀粉酶突变体或所述的宿主细胞的培养物的用途,用于水解糖苷键或用于形成简单糖;较佳地,所述的淀粉酶突变体用于水解α-1,4糖苷键;较佳地,所述的淀粉酶突变体用于水解淀粉或糖原中的α-1,4糖苷键。在本发明的另一方面,提供用于水解糖苷键或用于形成简单糖组合物,它含有:(a)所述的淀粉酶突变体或含有所述宿主细胞的培养物;以及(b)食品学或工业上可接受的载体。在本发明的另一方面,提供一种水解糖苷键或形成简单糖的方法,该方法包含:用所述的淀粉酶突变体处理待水解的底物。在一个优选例中,所述方法中,在ph4.5~7.5条件下,用所述的淀粉酶突变体处理待水解的底物;更佳地,在ph5~7条件下,用所述的淀粉酶突变体处理待水解的底物;和/或在温度50-90℃条件下,用所述的多肽处理待水解的底物;更佳地,在温度60-80℃条件下,用所述的多肽处理待水解的底物。在另一优选例中,所述的待水解的底物包括:淀粉,糖原,寡糖,纤维二糖。本发明的其它方面由于本文的公开内容,对本领域的技术人员而言是显而易见的。附图说明图1、od540对还原糖含量线性关系的标准曲线(葡萄糖为标准)。图2、本发明的淀粉酶突变体的半衰期测定。其中,a:70℃,b:85℃,c:95℃。图3、温度和ph值对组合突变酶的影响。具体实施方式本发明人经过深入的研究,在深海嗜热菌来源的淀粉酶gs4j-amya基础上,获得了淀粉酶突变体,所述突变体相对于深海嗜热菌来源的淀粉酶成熟肽,第181位和第182位氨基酸缺失;并且,第363位突变为甘氨酸或第463位突变为苏氨酸。本发明的淀粉酶突变体不仅具有原始淀粉酶的耐酸优势,而且热稳定性大大提高。如本文所用,所述的“简单糖”指多糖链的糖苷键被切割后形成的一类糖的总称,其链长度低于被切割前。例如,所述的简单糖含有1~50个葡萄糖,较佳的,含有1~40个葡萄糖;例如含有1~20个葡萄糖,如15、12、10、9、8、7、5、3、2、1个葡萄糖。所述的“简单糖”包括“葡萄糖”。如本文所用,所述的“葡萄糖”指一种含有六个碳原子的单糖。分子式c6h12o6。如本文所用,“分离的”是指物质从其原始环境中分离出来(如果是天然的物质,原始环境即是天然环境)。如活体细胞内的天然状态下的多聚核苷酸和多肽是没有分离纯化的,但同样的多聚核苷酸或多肽如从天然状态中同存在的其他物质中分开,则为分离纯化的。本发明中,所述的深海嗜热菌来源的淀粉酶gs4j-amya也称为野生型淀粉酶,其包含信号肽的核苷酸序列如seqidno:1所示(其中第1~102位为信号肽序列),氨基酸序列如seqidno:2所示;其不包含信号肽的成熟肽序列如seqidno:3所示。本发明两种优选的多肽分别命名为ig181-182*/c363g(第181位和第182位氨基酸缺失,第363位突变为甘氨酸)和ig181-182*n463t(第181位和第182位氨基酸缺失,第463位突变为苏氨酸)。本发明的多肽可以是重组多肽、合成多肽,优选重组多肽。本发明还包括ig181-182*/c363g和ig181-182*n463t多肽的片段、衍生物和类似物。如本文所用,术语“片段”、“衍生物”和“类似物”是指基本上保持本发明的天然ig181-182*/c363g和ig181-182*n463t多肽相同的生物学功能或活性的多肽。本发明的多肽片段、衍生物或类似物可以是(i)有一个或多个保守或非保守性氨基酸残基(优选保守性氨基酸残基)被取代的多肽,而这样的取代的氨基酸残基可以是也可以不是由遗传密码编码的,或(ii)在一个或多个氨基酸残基中具有取代基团的多肽,或(iii)成熟多肽与另一个化合物(比如延长多肽半衰期的化合物,例如聚乙二醇)融合所形成的多肽,或(iv)附加的氨基酸序列融合到此多肽序列而形成的多肽(如前导序列或分泌序列或用来纯化此多肽的序列或蛋白原序列,或与抗原igg片段的形成的融合蛋白)。根据本文的教导,这些片段、衍生物和类似物属于本领域熟练技术人员公知的范围。当然,所述的“片段”、“衍生物”和“类似物”在本文主张的突变位点上,氨基酸序列是保守的,也即相应于深海嗜热菌来源的淀粉酶成熟肽的第181位和第182位氨基酸缺失,第363位突变为甘氨酸或第463位突变为苏氨酸。在本发明中,术语“突变型淀粉酶多肽”包括具有与ig181-182*/c363g或ig181-182*n463t多肽相同功能的ig181-182*/c363g或ig181-182*n463t多肽的变异形式。这些变异形式包括(但并不限于):一个或多个(通常为1-50个,较佳地1-30个,更佳地1-20个,更佳地1-10个,最佳地1-5个)氨基酸的缺失、插入和/或取代,以及在c末端和/或n末端添加或缺失一个或数个(通常为20个以内,较佳地为10个以内,更佳地为5个以内)氨基酸。例如,在本领域中,用性能相近或相似的氨基酸进行取代时,通常不会改变蛋白质的功能。比如,在c末端和/或n末端添加或缺失一个或数个氨基酸通常也不会改变蛋白质的功能;又比如,仅表达该蛋白的催化结构域,而不表达碳水化合物结合结构域也能获得和完整蛋白同样的催化功能。因此该术语还包括ig181-182*/c363g或ig181-182*n463t多肽的活性片段和活性衍生物。一般地,所述变异形式发生在本文主张的突变位点之外,但是其仍然保留了ig181-182*/c363g或ig181-182*n463t多肽的活性。发明还提供ig181-182*/c363g或ig181-182*n463t多肽的类似物。这些类似物与ig181-182*/c363g或ig181-182*n463t多肽的差别可以是氨基酸序列上的差异,也可以是不影响序列的修饰形式上的差异,或者兼而有之。这些多肽包括天然或诱导的遗传变异体。诱导变异体可以通过各种技术得到,如通过辐射或暴露于诱变剂而产生随机诱变,还可通过定点诱变法或其他已知分子生物学的技术。类似物还包括具有不同于天然l-氨基酸的残基(如d-氨基酸)的类似物,以及具有非天然存在的或合成的氨基酸(如β、γ-氨基酸)的类似物。应理解,本发明的多肽并不限于上述例举的代表性的多肽。修饰(通常不改变一级结构)形式包括:体内或体外的多肽的化学衍生形式如乙酰化或羧基化。修饰还包括糖基化,如那些在多肽的合成和加工中或进一步加工步骤中进行糖基化修饰而产生的多肽。这种修饰可以通过将多肽暴露于进行糖基化的酶(如哺乳动物的糖基化酶或去糖基化酶)而完成。修饰形式还包括具有磷酸化氨基酸残基(如磷酸酪氨酸,磷酸丝氨酸,磷酸苏氨酸)的序列。还包括被修饰从而提高了其抗蛋白水解性能或优化了溶解性能的多肽。本发明的ig181-182*/c363g或ig181-182*n463t多肽的氨基端或羧基端还可含有一个或多个多肽片段,作为蛋白标签。任何合适的标签都可以用于本发明。例如,所述的标签可以是flag,ha,ha1,c-myc,poly-his,poly-arg,strep-tagii,au1,ee,t7,4a6,ε,b,ge以及ty1。这些标签可用于对蛋白进行纯化。为了使翻译的蛋白分泌表达(如分泌到细胞外),可以在所述ig181-182*/c363g或ig181-182*n463t多肽的氨基酸氨基末端加上引导多肽表达于细胞外的信号肽,或用引导多肽表达于细胞外的信号肽替换本身信号肽序列。信号肽在多肽从细胞内分泌出来的过程中可被切去。本发明的编码ig181-182*/c363g或ig181-182*n463t多肽的多核苷酸可以是dna形式或rna形式。dna形式包括cdna、基因组dna或人工合成的dna。dna可以是单链的或是双链的。术语“编码多肽的多核苷酸”可以是包括编码此多肽的多核苷酸,也可以是还包括附加编码和/或非编码序列的多核苷酸。本发明还涉及上述多核苷酸的变异体,其编码与本发明有相同的氨基酸序列的多肽或多肽的片段、类似物和衍生物。此多核苷酸的变异体可以是天然发生的等位变异体或非天然发生的变异体。这些核苷酸变异体包括取代变异体、缺失变异体和插入变异体。如本领域所知的,等位变异体是一个多核苷酸的替换形式,它可能是一个或多个核苷酸的取代、缺失或插入,但不会从实质上改变其编码的多肽的功能。本发明的编码ig181-182*/c363g或ig181-182*n463t多肽的多核苷酸的全长序列或其片段通常可以用pcr扩增法、重组法或人工合成的方法获得。对于pcr扩增法,可根据本发明所公开的有关核苷酸序列,尤其是开放阅读框序列来设计引物,并用市售的cdna库或按本领域技术人员已知的常规方法所制备的cdna库作为模板,扩增而得有关序列。当序列较长时,常常需要进行两次或多次pcr扩增,然后再将各次扩增出的片段按正确次序拼接在一起。一旦获得了有关的序列,就可以用重组法来大批量地获得有关序列。这通常是将其克隆入载体,再转入细胞,然后通过常规方法从增殖后的宿主细胞中分离得到有关序列。此外,还可用人工合成的方法来合成有关序列,尤其是片段长度较短时。通常,通过先合成多个小片段,然后再进行连接可获得序列很长的片段。目前,已经可以完全通过化学合成来得到编码本发明蛋白(或其片段,或其衍生物)的dna序列。然后可将该dna序列引入本领域中已知的各种现有的dna分子(或如载体)和细胞中。此外,还可通过化学合成将突变引入本发明蛋白序列中。本发明也涉及包含本发明的多核苷酸的载体,以及用本发明的载体或ig181-182*/c363g或ig181-182*n463t多肽编码序列经基因工程产生的宿主细胞,以及经重组技术产生本发明所述多肽的方法。本发明中,编码ig181-182*/c363g或ig181-182*n463t多肽的多核苷酸序列可插入到重组表达载体中。术语“重组表达载体”指本领域熟知的细菌质粒、噬菌体、酵母质粒、植物细胞病毒、哺乳动物细胞病毒或其他载体。总之,只要能在宿主体内复制和稳定,任何质粒和载体都可以用。表达载体的一个重要特征是通常含有复制起点、启动子、标记基因和翻译控制元件。本领域的技术人员熟知的方法能用于构建含编码ig181-182*/c363g或ig181-182*n463t多肽的dna序列和合适的转录/翻译控制信号的表达载体。这些方法包括体外重组dna技术、dna合成技术、体内重组技术等。所述的dna序列可有效连接到表达载体中的适当启动子上,以指导mrna合成。表达载体还包括翻译起始用的核糖体结合位点和转录终止子。此外,表达载体优选地包含一个或多个选择性标记基因,以提供用于选择转化的宿主细胞的表型性状,如真核细胞培养用的二氢叶酸还原酶、新霉素抗性以及绿色荧光蛋白(gfp),或用于大肠杆菌的卡那霉素或氨苄青霉素抗性。包含上述的适当dna序列以及适当启动子或者控制序列的载体,可以用于转化适当的宿主细胞,以使其能够表达蛋白质。宿主细胞可以是原核细胞,如细菌细胞;或是低等真核细胞,如酵母细胞;或是高等真核细胞,如植物细胞。代表性例子有:大肠杆菌,链霉菌属、农杆菌;真菌细胞如酵母;植物细胞等。本发明的多核苷酸在高等真核细胞中表达时,如果在载体中插入增强子序列时将会使转录得到增强。增强子是dna的顺式作用因子,通常大约有10到300个碱基对,作用于启动子以增强基因的转录。本领域一般技术人员都清楚如何选择适当的载体、启动子、增强子和宿主细胞。用重组dna转化宿主细胞可用本领域技术人员熟知的常规技术进行。获得的转化子可以用常规方法培养,表达本发明的基因所编码的多肽。根据所用的宿主细胞,培养中所用的培养基可选自各种常规培养基。在适于宿主细胞生长的条件下进行培养。当宿主细胞生长到适当的细胞密度后,用合适的方法(如温度转换或化学诱导)诱导选择的启动子,将细胞再培养一段时间。通过常规的重组dna技术,利用本发明的多聚核苷酸序列来表达或生产重组的ig181-182*/c363g或ig181-182*n463t多肽,一般来说有以下步骤:(1).用本发明的编码ig181-182*/c363g或ig181-182*n463t多肽的多核苷酸(或变异体),或用含有该多核苷酸的重组表达载体转化或转导合适的宿主细胞;(2).在合适的培养基中培养的宿主细胞;(3).从培养基或细胞中分离、纯化蛋白质。在本发明的一个具体实施例中,通过设计突变引物,以构建好的含有gs4j-amya野生型基因序列的pet-28a质粒为模板,进行全质粒扩增点突变,将ig两个氨基酸缺失,然后继续突变c363g和n463t,获得含有突变淀粉酶基因的pet-28a重组载体。之后,将重组载体转化大肠杆菌,诱导表达,获得所述的淀粉酶突变体。在上面的方法中的重组多肽可在细胞内、或在细胞膜上表达、或分泌到细胞外。如果需要,可利用其物理的、化学的和其它特性通过各种分离方法分离和纯化重组的蛋白。这些方法是本领域技术人员所熟知的。这些方法的例子包括但并不限于:常规的复性处理、用蛋白沉淀剂处理(盐析方法)、离心、渗透破菌、超处理、超离心、分子筛层析(凝胶过滤)、吸附层析、离子交换层析、高效液相层析(hplc)和其它各种液相层析技术及这些方法的结合。本发明还提供了一种组合物,它含有有效量的本发明的ig181-182*/c363g或ig181-182*n463t多肽以及食品学上或工业上可接受的载体或赋形剂。这类载体包括(但并不限于):水、缓冲液、葡萄糖、水、甘油、乙醇、及其组合。本领域技术人员可根据组合物的实际用途确定组合物中ig181-182*/c363g或ig181-182*n463t多肽的有效量。所述的组合物中还可添加调节本发明的ig181-182*/c363g或ig181-182*n463t多肽酶活性的物质,例如一些金属离子的添加可以增加或降低酶的活性。任何具有提高酶活性功能的物质均是可用的。本领域技术人员可根据组合物的实际用途确定添加改善酶活特性的物质。本发明的ig181-182*/c363g或ig181-182*n463t可作用于水解糖苷键,形成简单糖。在获得了本发明的ig181-182*/c363g或ig181-182*n463t后,根据本发明的提示,本领域人员可以方便地应用该酶来发挥水解底物的作用。作为本发明的优选方式,还提供了一种形成简单糖的方法,该方法包含:用本发明所述的ig181-182*/c363g或ig181-182*n463t处理待水解的底物,所述的底物包括淀粉、糖原、寡糖、纤维二糖,以及它们的混合物。通常,在ph4.5~7.5、优选为ph5~7条件下,用所述的ig181-182*/c363g或ig181-182*n463t处理待水解的底物。通常在50-90℃、优选为60-80℃条件下,用所述的ig181-182*/c363g或ig181-182*n463t处理待水解的底物。本发明的多肽对于糖苷键具有识别和水解作用,因此,一系列糖链中具有糖苷键的物质均可作为本发明的多肽的底物。这些具有糖苷键的物质包括但不限于:淀粉、糖原、寡糖、纤维二糖等,或它们的混合物。本发明的多肽水解糖苷键的位置在α-1,4糖苷键。本发明所述的淀粉酶突变体ig181-182*/c363g和ig181-182*/n463t,在85℃的半衰期分别由对照(突变前)的5.3min提高到77.0min和82.5min。相对于筛菌或诱变等非理性的方法,缩短了酶学性质改造时间。将该热稳定性好的酸性淀粉酶突变体应用于食品、医药、化工等领域,可以在酸性和高温环境中高效降解淀粉酶,具有广阔的应用前景。下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件如j.萨姆布鲁克等编著,分子克隆实验指南,第三版,科学出版社,2002中所述的条件,或按照制造厂商所建议的条件。实施例1、淀粉酶热稳定性定点突变针对bla(bacilluslecheniformisα-淀粉酶)和bsta(bacillusstearothermophilusα-淀粉酶),本发明人进行了广泛的研究,探寻其中与热稳定性相关的位点。经过深入研究比对,本发明人找到酸性α-淀粉酶gs4j-amya中相对应的位点,把相应的位点突变成热稳定性较好的氨基酸残基来提高本实验中酸性α-淀粉酶gs4j-amya的热稳定性。所述的淀粉酶ig181-182*/c363g的成熟肽,相对于野生型的深海嗜热菌来源的淀粉酶gs4j-amya成熟肽(seqidno:3)序列,缺失了第181位和第182位两个氨基酸,并且在相应于野生型序列的第363位用甘氨酸替换了半胱氨酸。所述的淀粉酶ig181-182*/n463t的成熟肽,相对于野生型的深海嗜热菌来源的淀粉酶gs4j-amya成熟肽(seqidno:3),缺失了第181位和第182位两个氨基酸,并且在相应于野生型序列的第463位用苏氨酸替换了天冬酰胺。1、突变α-淀粉酶基因的获得(1)引物设计以突变位点为中心前后各16-20bp,正反向引物是互补的,引物总长40bp左右,gc含量在40%以上。引物如表2。表2、突变α-淀粉酶引物*以加黑下划线标示的碱基为突变位点的碱基。(2)pcr扩增根据得到的高温淀粉酶基因amya全序列(seqidno:1)设计如下引物:上游引物am-oa-f:5’-agcgagctccaccaccaccaccaccacatgaaaaagacagctatcgcgattgcagtggcactggctggtttcgctaccgtagcgcaggcccaccaccaccaccaccacgccgcaccgtttaacg-3’(seqidno:4);下游引物am-oa-r:cccgctcgaggtggtggtggtggtggtggtggtggtggtggtggtgaggccatgccaccaac(seqidno:5)。其中,上游引物中包含了大肠杆菌外膜蛋白信号肽ompa的编码序列。用pcr扩增得到高温淀粉酶基因amya全序列。该pcr条件为95℃预变性5min,随后95℃30s、48℃30s、72℃1min进行32个循环,最后,72℃延伸10min。琼脂糖凝胶电泳显示1700bp左右处有一特异性条带,将其从琼脂糖凝胶上切下,用scai和xhoi酶切,琼脂糖凝胶电泳回收1700bp左右的dna片段。将大肠表达载体pet-28a同样用scai和xhoi酶切,琼脂糖凝胶电泳回收5300bp的dna片段,将其与上述所得1700bp的的dna片段连接后,按照标准的氯化钙法转化入大肠杆菌top10中,筛选具有硫酸卡那霉素抗性的转化子。lb液体培养基(含硫酸卡那霉素)中培养后使用t7通用引物进行pcr验证,pcr验证程序为95℃预变性5min,随后95℃30s、48℃30s、72℃1min进行30个循环,最后在72℃延伸10min。琼脂糖凝胶电泳显示在2000bp左右有正确条带的为阳性转化子,将对应的正确的菌株送测序,测序显示序列正确,质粒构建正确。将该阳性转化子命名为pet28a-amyap/top10,含有的质粒命名为pet28a-amyap。以pet28a-amyap质粒为模板,进行pcr扩增:50μl或25μlpcr反应体系皆可,以50μl为例,体系中包括25μl高保真dna聚合酶primestarmax(2×)、带有突变位点的正向和反向引物各1μl,加入0.1~10ng质粒模板,根据模板加入的体积调整ddh2o加入体积。pcr反应程序为:95℃预变性5min、98℃变性10s、tm-3℃退火5s、72℃延伸,22个延伸循环后,最终72℃延伸1min。反应结束后,取pcr产物5μl,用0.8%的琼脂糖凝胶电泳检测是否有pcr扩增产物。(3)dpni酶消化模板dpni酶消化模板,10μl反应体系中包括1μl10×nebcutsmartbuffer、8.75μlpcr产物和0.25μldpni酶液。10μl混合液置于37℃水浴,反应5~6h。(4)转化去除模板的10μl的pcr产物转化大肠杆菌top10,涂含有卡那霉素的lb固体平板来筛选转化子,培养12~16h,挑取单菌落,菌落pcr验证正确后,将菌液送公司测序,验证目的位点是否突变成功。从测序正确的大肠杆菌top10菌株中抽提突变后的质粒,取3~5μl转化e.colibl21(de3)plyss菌株,得到突变酶的表达菌株。2、突变酶在大肠杆菌中诱导表达(1)从lb平板上挑取大肠杆菌单菌落或接种1%甘油菌,接种于3~5ml含有50μg/ml卡那霉素的lb液体培养基中,37℃、200r/min培养12h左右,作为一级种。(2)将一级种按2%(v/v)接种量接种到加有100μl卡那霉素(50mg/ml)的100mllb液体培养基中,37℃、200r/min条件下培养2h左右,测得od600=0.6~0.8时,加入100μliptg(1mol/l)至100ml培养液中诱导(iptg终浓度为1mmol/l),37℃,200r/min条件下诱导48h。(3)诱导36h时,取样1ml置于ep管中,8000g离心1min,取上清用于测定淀粉酶酶活,检测是否表达成功。结果表明,成功地获得了表达突变酶ig181-182*/c363g和ig181-182*/n463t的重组菌株,成功诱导表达获得了突变酶ig181-182*/c363g和ig181-182*/n463t。同时,本发明人采用相同的方案,也制备了表达野生型gs4j-amya酶的重组菌株,表达了野生型gs4j-amya酶。实施例2、淀粉酶的大肠杆菌异源表达与纯化1、野生型酶和突变酶在大肠杆菌中诱导表达(1)从lb平板上分别挑取表达野生型酶和突变酶的大肠杆菌单菌落或接种1%甘油菌,接种于3~5ml含有50μg/ml卡那霉素的lb液体培养基中,37℃、200r/min培养12h左右,作为一级种。(2)将一级种按2%(v/v)接种量接种到加有100μl卡那霉素(50mg/ml)的100mllb液体培养基中,37℃、200r/min条件下培养2h左右,测得od600=0.6~0.8时,加入100μliptg(1mol/l)至100ml培养液中诱导(iptg终浓度为1mmol/l),37℃,200r/min条件下诱导48h。2、淀粉酶纯化(1)收集诱导表达获得的发酵液上清所构建的质粒带有大肠杆菌外膜蛋白ompa信号肽,所以重组e.coli菌体将淀粉酶分泌到胞外。每个突变酶菌株诱导500ml,诱导48h后,将发酵液5000g离心20min,收集上清。(2)浓缩并换液用millipore公司的实验室规模的切向流超滤装置(截留分子量为10kda的超滤膜包)超滤浓缩,第一次将样品浓缩至50ml左右,然后加入450ml左右的缓冲液(ph7.4,10mmol/lnah2po4,10mmol/lna2hpo4,0.5mol/lnacl,20mmol/l咪唑(不同突变酶所用的咪唑浓度不同),进行第二次浓缩,将样品从发酵液换成含咪唑的平衡缓冲液a,最终浓缩了10倍左右。(3)亲和层析首先用超纯水(使用前超声15min左右去除其中气泡)将镍柱中的乙醇冲洗干净,1ml/min的流速,15min左右。然后用平衡缓冲液a平衡镍柱,1ml/min,10min,接着进样,结束后用平衡缓冲液a洗杂,15~20min,用含250mmol/l咪唑的缓冲液洗脱目的蛋白,收集洗脱峰,每管1ml,用nanodrop测定每个收集管中的蛋白浓度,弃掉无蛋白的收集管,取一管或几管蛋白浓度较高的收集组分进行sds-page检测纯度。(4)保存镍柱用500mmol/l咪唑的缓冲液冲洗,将所有蛋白彻底洗脱下来,1ml/min,10min,然后用超纯水洗镍柱,10min左右,最后用20%的乙醇液封镍柱,4℃保存。(5)超滤浓缩并去除咪唑将收集到的淀粉酶组分转移到超滤管(截留分子量为10kda)中,5000g,离心20min,再加入15mlpbs(ph7.2~7.4),混匀,继续离心,这样重复两次,最终将蛋白样品浓缩至1ml左右,并用nanodrop测蛋白浓度,观察吸收峰(咪唑在230nm波长处有很强吸收峰),初步检测咪唑去除程度。3、蛋白sds-page方法采用12%分离胶,5%浓缩胶。浓缩胶90v,电泳30min,接着分离胶120v,电泳70min,用配制好的考马斯亮蓝r-250试剂室温染色过夜,然后进行脱色,2h换一次脱色液,换2-3次基本可以看到清晰的蛋白条带,用凝胶成像仪拍照记录。实施例3、淀粉酶酶活力测定方法1、dns试剂测还原糖含量标准曲线的制作(1)用无水葡萄糖配制1mg/ml的葡萄糖溶液:称取大于1g的无水葡萄糖在烘箱里65℃烘干,准确称取1.000g无水葡萄糖于烧杯中,用少量去离子水溶解,转移至1l的容量瓶中,准确定容到1l,即得1mg/ml的葡萄糖溶液。(2)按表3加样,沸水浴5min,冰水浴中冷却终止反应,加8ml去离子水,定容到10ml,混匀,室温放置20min左右,测定540nm波长处吸光值,绘制标准曲线。获得的标准曲线如图1。表3还原糖标准曲线绘制加样体系2、酶活测定定义:在酶促反应条件下,1min催化可溶性淀粉底物生成1μmol还原糖所需α-淀粉酶的酶量为一个酶活单位(u)。根据dns试剂测量葡萄糖含量的标准曲线(图1),计算单位体积的淀粉酶酶活:3、比酶活测定比酶活测定是通过测定纯酶的酶活(u/ml)和纯酶的蛋白浓度(mg/ml),然后计算酶活和蛋白浓度的比值,即得比酶活(u/mg)。测得的突变型酶ig181-182*/c363g和ig181-182*/n463t的比酶活如表3所示。表4酶淀粉酶比酶活ig181-182*/c363g10085.3u/mgig181-182*/n463t15345.7u/mg由表4的结果可见,突变型的酶具有良好的比酶活。实施例4、野生型和突变型淀粉酶在高温条件下半衰期的测定将纯化得到的酶液(ph7.2~7.4)用ph5.5(25mmol/l醋酸-醋酸钠缓冲液)稀释到合适倍数(稀释到5-13u/ml,尽量使得测定的od540在0.2-0.8范围内),70℃水浴孵育,每隔一段时间取样,与可溶性淀粉底物进行酶促反应,测定残存的酶活。将稀释好的酶液分装成30μl每管,置于pcr仪中,85℃和95℃条件下处理不同时间,测定残存酶活。根据恒定温度和ph值、底物浓度无关的酶失活的一级动力学方程为:或其中kd为酶失活常数,t表示热处理时间,e0和et分别指初始和时间t时的相对酶活。kd可通过ln(et/e0)对时间t作曲线的斜率求得,然后半衰期t1/2即为ln2与k的比值:测得的各酶的在高温下的半衰期情况如图2所示。由图2的结果可见,不管在70℃,85℃还是95℃,突变型酶ig181-182*/c363g和ig181-182*/n463t的半衰与野生型酶均发生了极其显著的延长。以85℃为例,ig181-182*/c363g和ig181-182*/n463t的半衰期分别由野生型的5.3min提高到77.0min和82.5min。上述结果表明,突变型酶在高温下的热稳定性发生了显著性提高。实施例5、突变型淀粉酶的最适温度测定用ph5.5(25mmol/l醋酸-醋酸钠缓冲液)的缓冲液配制1%(w/v)淀粉底物,在不同温度条件下测定酶活力,然后将酶活力最高值设为100%,计算其余温度下的酶活力与最高值的百分比。结果如图3a,突变型酶与野生型酶的最适温度均在70℃左右。在50~90℃均保留较好的酶活性。实施例6、突变型淀粉酶的最适ph测定分别选用两种缓冲体系25mmol/lhac-naac缓冲体系(ph3~5.8)和25mmol/lna2hpo4-nah2po4(ph5.8~8.0)分别配制1%的可溶性淀粉底物,然后在最适温度条件下测定每个突变α-淀粉酶在不同ph时的酶活力,选ph3~5.8范围内最高的酶活定义为100%,计算出其余的ph条件下的淀粉酶酶活与最高酶活的百分比。结果如图3b,突变型酶与野生型酶的最适温度均在约ph5~7。在ph4.5~7.5时,均保留较好的酶活性。该结果也表明,突变型淀粉酶适用的ph范围宽,具有良好的耐酸性。在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1