本发明涉及一种可用于光响应聚合物制备的链转移剂,具体涉及的是一种光致变色链转移剂的制备方法及其应用。
背景技术:
1998年,Rizzardo在第37届国际高分子大会上首次提出了可逆加成-断裂链转移自由基聚合(RAFT)的概念。RAFT聚合方法具有以下优点:(1)适用的单体范围广。除了常见单体甲基丙烯酸甲酯和苯乙烯外,丙烯酸、对乙烯基苯磺酸钠、甲基丙烯酸羟乙酯、甲基丙烯酸胺基乙酯等特殊单体均可顺利聚合,有利于含特殊官能团烯类单体的聚合反应。(2)可用于合成多种拓扑型结构共聚物和均聚物。(3)聚合反应温度相对较低(60-70摄氏度)。(4)合成的聚合物的分子量可控,分子量分布较窄。目前常用的RAFT链转移剂有四种:二硫代酯类;三硫代碳酸酯类;黄原酸酯类;二硫代氨基甲酸酯类。与其中三种相比,三硫代碳酸酯类链转移剂较稳定,高温下不易分解,且易于合成。更为重要的是,使用三硫代碳酸酯类链转移剂可以避免阻滞效应。
在众多高分子材料中,光致变色材料是一类可用于涂料工业、防护伪装、光电信息技术、生物和化学传感等领域的新材料。光致变色材料按照化学成分划分可分别无机材料和有机材料两类,由于无机材料通常含有重金属元素或者稀土元素,且不易进行分子修饰,所以限制了其更进一步的应用。与之相比较,基于有机高分子的光致变色材料受到了广泛的关注,如甲亚胺类高分子,偶氮苯类高分子,螺吡喃类高分子和二芳杂环基乙烯类高分子等。其中螺吡喃类高分子有良好的着色能力和抗光疲劳性能。制备光致变色螺吡喃高分子通常有两种途径,一种是通过共聚或者接枝反应,将光致变色螺吡喃单元连接到聚合物的主链或者侧链上,这种方法的不足之处是合成较为复杂,螺吡喃的引入较为困难。另一种是把光致变色螺吡喃与其它材料共混使新材料具有光致变色性能,这种方法的不足之处是螺吡喃在材料的长期使用过程容易出现泄漏。
本发明采用酯化反应的方式,将螺吡喃类化合物和三硫代碳酸酯类链转移剂结合起来,得到一种新型光致变色链转移剂。再将这种链转移剂应用于苯乙烯的RAFT聚合中,可得到一种具有良好光致变色性能的聚合物。结果表明,使用这种链转移剂制备的聚合物不仅具有良好的光致变色性能,而且分子量可调控,分子量分布较窄,显示出较好的可调控性。
技术实现要素:
本发明的目的在于提供一种光致变色链转移剂、制备方法及其应用。该光致变色链转移剂的制备方法简单,将光致变色化合物:螺吡喃类化合物和链转移剂:三硫代碳酸酯类链转移剂通过酯化反应结合起来,形成一种具有光致变色功能的链转移剂,缩写为SPTTC。
一种光致变色链转移剂的制备方法,具体步骤如下:
(1)将螺吡喃类化合物和三硫代碳酸酯类链转移剂和4-二甲氨基吡啶加入干燥的二氯甲烷,在避光和冰浴条件下搅拌0.5-1小时。
(2)将含二环己基碳二亚胺的二氯甲烷溶液缓慢滴加到上述溶液中,滴加完成后在室温下避光反应24小时。
(3)将所得到的溶液浓缩后用硅胶柱进一步提纯,干燥后得到具有光致变色功能的链转移剂SPTTC。
上述制备方法中所用螺吡喃类化合物上的取代基R1为H,Br,Cl,甲氧基,硝基中的一种;三硫代碳酸酯类链转移剂上的取代基R2为C2-C17的正烷基中的一种。
上述制备方法中所用螺吡喃类化合物、三硫代碳酸酯类链转移剂、4-二甲氨基吡啶和二环己基碳二亚胺的摩尔比为:1:1~2:0.1~1:1~2,优选为1:1.1~1.3:0.2~0.4:1.1~1.3。
光致变色链转移剂在光致变色聚合物合成中的应用,具体步骤如下:
(1)以SPTTC为链转移剂,偶氮二异丁腈为引发剂,苯乙烯为单体,进行可逆加成断裂链转移自由基聚合,温度为70~120度,优选为90度,反应时间为15~75分钟。反应完成后,用四氢呋喃稀释反应物,并在甲醇中沉淀,再离心后干燥得到淡黄色聚合物固体。
(2)上述应用中,所用偶氮二异丁腈,SPTTC和苯乙烯的摩尔比为:1:2~10:100~500,优选为1:3~5:200~300。
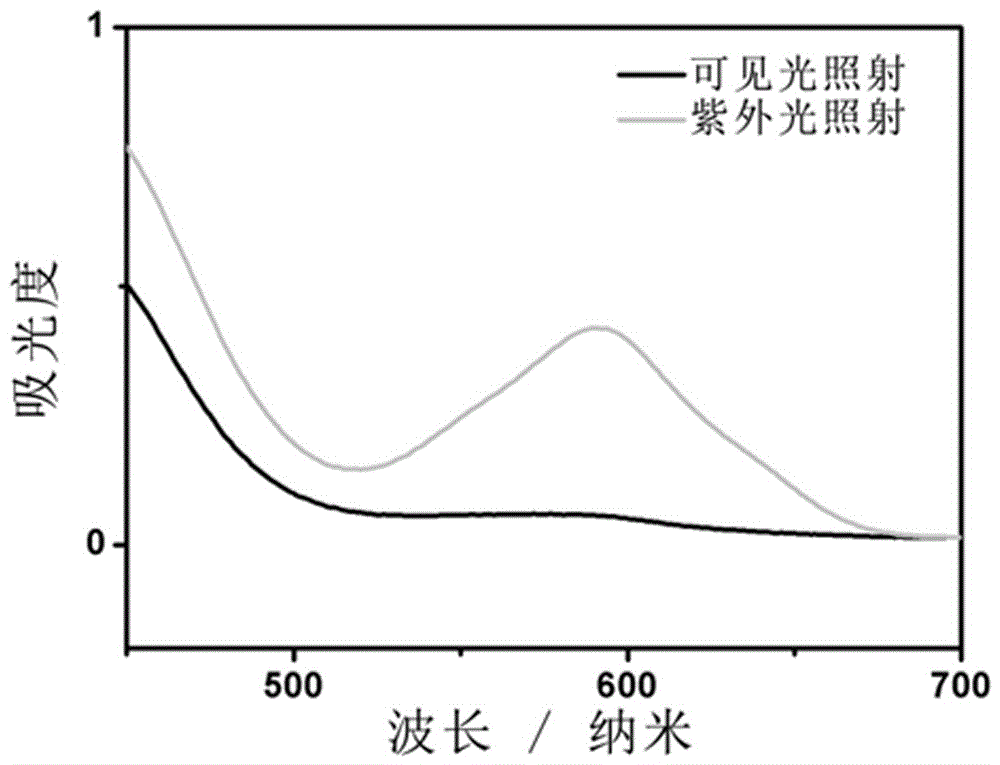
本发明通过酯化反应的方式,将光致变色化合物螺吡喃类化合物和三硫代碳酸酯类链转移剂结合起来,形成一种具有光致变色性能的链转移剂。再将这种链转移剂使用到苯乙烯的RAFT聚合中,得到具有光致变色性能的聚合物。将所得聚合物溶解在有机溶剂中如四氢呋喃,测试其在可见光和紫外光照射后的吸收光谱和荧光光谱。在可见光照射条件下,样品溶液在580nm处几乎没有吸收峰,在紫外光照射条件下,样品溶液在580nm处出现了强的吸收峰。再以560nm作为激发波长,样品溶液在可见光照射后没有出现荧光峰,在紫外光照射后于630nm处出现荧光峰。
本发明提供的光致变色链转移剂制备简单,光致变色性能好,易于制备具有良好光致变色性能的聚合物,且工艺简单,后处理方便,有利于大规模工业化生产,有望在功能材料科学领域得到广泛应用。
附图说明
图1为实施例1所制备的光致变色链转移剂在CDCl3中的核磁表征的氢谱图。
图2为实施例1所制备的光致变色链转移剂在四氢呋喃中的紫外吸收光谱图。
图3为实施例1所制备的光致变色链转移剂在四氢呋喃中的荧光发射光谱图。
图4为不同反应时间的聚合物的GPC曲线图。
图5为实施例5所制备的聚合物溶液的紫外吸收光谱图。
图6为实施例5所制备的聚合物溶液的荧光发射光谱图。
图7为实施例9所制备的聚合物膜在可见光照射后的照片。
图8为实施例9所制备的聚合物膜在紫外光照射后的照片。
具体实施方式
下面结合附图及具体实施例对本发明作进一步详细说明。
实施例1:一种光致变色链转移剂的制备方法,具体步骤如下:
将N-羟乙基-3,3-二甲基-6'-硝基吲哚啉螺吡喃100mg(0.28mmol), 2-(十二烷基三硫代碳酸酯基)-2-异丁酸112mg(0.31mmol)和4-二甲氨基吡啶7mg(0.06mmol )加入到25ml两口圆底烧瓶中,再加入5ml干燥的二氯甲烷搅拌溶解,之后在避光和冰浴条件下搅拌0.5小时。
(2)将75mg(0.33mmol)的二环己基碳二亚胺加入到5ml干燥的二氯甲烷中溶解,随后转移到恒压滴液漏斗中。
(3)将步骤(2)中所得的溶液缓慢滴加到步骤(1)所得到的溶液中,滴加完成后在室温下避光反应24个小时。
(4)对反应后的混合液体抽滤,去除生成的白色固体。将所得到的溶液浓缩后用柱层析色谱法进一步提纯,所用展开剂为二氯甲烷,干燥后得到暗黄色固体,2-(十二烷基三硫代碳酸酯基)-2-异丁酸-3,3-二甲基-6'-硝基吲哚啉螺吡喃-N-乙酯,产率为74%。产物结构以核磁共振氢谱分析,所用溶剂为氘代氯仿,结果如图1所示,将产物溶于四氢呋喃,并测试其在紫外光和可见光照射后的光致变色和荧光性能,结果如图2和图3所示。
实施例2:一种光致变色链转移剂的制备方法,具体步骤如下:
将N-羟乙基-3,3-二甲基-5-氯-6'-硝基吲哚啉螺吡喃110mg(0.28mmol), 2-(十二烷基三硫代碳酸酯基)-2-异丁酸120mg(0.33mmol)和4-二甲氨基吡啶13mg(0.11mmol)加入到25ml两口圆底烧瓶中,再加入5ml干燥的二氯甲烷搅拌溶解,之后在避光和冰浴条件下搅拌0.5小时。
(2)将70mg(0.31mmol)的二环己基碳二亚胺加入到5ml干燥的二氯甲烷中溶解,随后转移到恒压滴液漏斗中。
(3)将步骤(2)中所得的溶液缓慢滴加到步骤(1)所得到的溶液中,滴加完成后在室温下避光反应24个小时。
(4)对反应后的混合液体抽滤,去除生成的白色固体。将所得到的溶液浓缩后用柱层析色谱法进一步提纯,所用展开剂为二氯甲烷,干燥后得到暗黄色固体,2-(十二烷基三硫代碳酸酯基)-2-异丁酸-3,3-二甲基-5-氯-6'-硝基吲哚啉螺吡喃-N-乙酯,产率为78%。
实施例3:一种光致变色链转移剂的制备方法,具体步骤如下:
将N-羟乙基-3,3-二甲基-6'-硝基吲哚啉螺吡喃100mg (0.28mmol), 2-(十四烷基三硫代碳酸酯基)-2-异丁酸137mg(0.36mmol)和4-二甲氨基吡啶9mg(0.08mmol) 、加入到25ml两口圆底烧瓶中,再加入5ml干燥的二氯甲烷搅拌溶解,之后在避光和冰浴条件下搅拌0.5小时。
(2)将81mg(0.36mmol)的二环己基碳二亚胺加入到5ml干燥的二氯甲烷中溶解,随后转移到恒压滴液漏斗中。
(3)将步骤(2)中所得的溶液缓慢滴加到步骤(1)所得到的溶液中,滴加完成后在室温下避光反应24个小时。
(4)对反应后的混合液体抽滤,去除生成的白色固体。将所得到的溶液浓缩后用柱层析色谱法进一步提纯,所用展开剂为二氯甲烷,干燥后得到暗黄色固体,2-(十四烷基三硫代碳酸酯基)-2-异丁酸-3,3-二甲基-6'-硝基吲哚啉螺吡喃-N-乙酯,产率为76%。
实施例4:一种光致变色链转移剂在四氢呋喃溶液中的光谱性质
取1mg实施例1所制备的光致变色链转移剂SPTTC于5ml比色皿中,加入3ml四氢呋喃溶解,测定其在可见光和紫外光照射后的吸收光谱,见图2。再以560nm作为激发波长,测定其在可见光和紫外光照射后的荧光光谱,见图3。
实施例5:一种具有光致变色性能的聚合物的制备方法,具体步骤如下:
(1)将实施例1制备得到的光致变色链转移剂和引发剂偶氮二异丁腈按照质量分别为104mg(0.15mmol)、8mg(0.05mmol)加入到10ml交换反应管中,再加入1500mg(14.42mmol)的苯乙烯溶解完全。
(2)在冰浴条件下,将反应管抽真空-充氮气循环3次。
(3)将反应管置于90摄氏度下反应15-75分钟,反应完成后,将反应管置于冰浴中以停止反应。
(4)用四氢呋喃稀释反应物,并在甲醇中沉淀,再离心后干燥得到淡黄色固体。
实施例6:一种具有光致变色性能的聚合物的制备方法,具体步骤如下:
(1)将实施例1制备得到的光致变色链转移剂和引发剂偶氮二异丁腈按照质量分别为173mg(0.25mmol)、8mg(0.05mmol)加入到10ml交换反应管中,再加入1040mg(10.00mmol)的苯乙烯溶解完全。
(2)在冰浴条件下,将反应管抽真空-充氮气循环3次。
(3)将反应管置于90摄氏度下反应15-75分钟,反应完成后,将反应管置于冰浴中以停止反应。
(4)用四氢呋喃稀释反应物,并在甲醇中沉淀,再离心后干燥得到淡黄色固体。
实施例7:具有光致变色性能的聚合物的GPC测试
取实施例5所制备的样品4mg于5ml干净小管中,加入2ml色谱级四氢呋喃溶解,再用0.22微米规格的微孔过滤器过滤,将所制得的聚合物溶液进行GPC测试,结果如图4。测试结果表明:该聚合物样品具有可调控的分子量和较窄的分子量分布。
实施例8:具有光致变色性能的聚合物的四氢呋喃溶液的光谱性质
取实施例5所制备的聚合物3mg到5ml的比色皿中,用3ml四氢呋喃溶解,测定其在可见光和紫外光照射后的吸收光谱,见图5。再以560nm为激发波长,测定其在可见光和紫外光分别照射后的荧光图谱,见图6。测试结果表明:实施例5所制备的聚合物具有光致变色性能。
实施例9:具有光致变色性能的聚合物膜的制备
取实施例5制备的聚合物固体溶于少量氯苯,质量分数优选为5%,并用真空旋涂机将其均匀的涂覆在单面抛光的硅片上,转速为第一阶段1500转/分,第二阶段3500转/分。得到的膜片在可见光照射后的照片如图7所示,在可见光照射后,膜片为无色透明,且没有红色荧光。在紫外光照射后的照片如图8所示,在紫外光照射后,膜片为紫红色,且有红色荧光。此外,该膜片经可见光再次照射后又可以回复到没有颜色和荧光的状态,且该可程可实现多次可逆。结果表明:实施例9所制备的聚合物膜具有良好的光致变色性能。
上述实施例用来解释说明本发明,而非对本发明进行限制,在本发明的精神和权利要求的保护范围内,对本发明所做出的任何修改和改变,都落入本发明的保护范围。