一套将质粒表达系统经优化重组到大肠杆菌染色体的方法与流程
本发明涉及生物技术和合成生物学领域,具体地说,涉及一套将质粒上的表达系统经过优化,无痕插入染色体从而得到完全替代原质粒功能的重组大肠杆菌构建方法。。
背景技术:
大肠杆菌是科学研究中重要的模式生物,为了研究其生命活动,人们借助许多外源系统来开发研究手段和工具,导入大肠杆菌体内进行活细胞的研究。例如Lei Wang,Peter G Schultz等人开发了遗传密码扩展技术,将一对正交的氨酰tRNA合成酶-tRNA引入大肠杆菌中,使其可以识别人工合成的具有特定功能的非天然氨基酸,并将其插入目标蛋白质的特定位置,来研究体内蛋白质的功能和对目标蛋白质进行修饰调控。在合成生物学领域,人们也会向大肠杆菌体内导入一些具有特定功能的控制元件(通常是蛋白质或者核酸),得到具有特殊功能的菌体。同样在工业生产中,大肠杆菌是重要的生产载体,导入可表达的外源基因后作为宿主细胞,可以生产大量的生物制剂,如重组人胰岛素、酶类等。
因此向大肠杆菌体内导入外源系统有非常广泛的用途,然而目前基本都是依靠于质粒载体进行所需外源基因的表达,不仅需要加入抗生素进行质粒的筛选和维持,同时由于质粒复制子类型和筛选抗性类型的限制,多质粒无法共存,给研究带来了很多不便。为了不依赖于质粒载体,外源基因可以通过重组至大肠杆菌染色体上而稳定的存在于菌体内。但如果外源基因在细菌染色体上插入的位置不当,不仅可能会造成外源系统欠表达或过表达,同时也会造成某些内源蛋白质或者核酸的表达缺陷,干扰大肠杆菌宿主的正常生长。
现有技术常选择非必需的基因或者某些目前未知功能的基因位置进行插入替换,例如在大肠杆菌代谢乳糖的lacZ基因处插入木聚糖酶,或者在未知功能的基因yihP处插入蔗糖代谢酶等等。因此内源的基因表达会被破坏,菌体会缺失原本正常表达出来的某种蛋白质,尽管在一般研究所使用的培养条件下,并未出现严重的生长障碍,但因缺乏充分的检查和验证,很可能菌株存在潜在的生长障碍和代谢问题,会在某种实际应用的条件下突显出来,从而阻碍科学研究或者工业生产。此外,重组时如果留下筛选标记(通常是抗性基因),菌体会有潜在的抗生素抗性扩散危险,并且残留的无关核酸序列会限制菌体进一步的重组改造。
因此,亟需研发一种将有价值的基因从现有多拷贝质粒转移到大肠杆菌染色体的方法,包括:无痕的重组大肠杆菌构建方法;经过测试不影响大肠杆菌菌体生长和代谢的染色体重组位置;合适的启动子和终止子序列使其在单拷贝基因组上的表达量能与多拷贝质粒相当。
技术实现要素:
为了解决现有技术中存在的问题,本发明的目的是提供一种构建染色体上插入外源表达系统的大肠杆菌的方法,可以完全替代使用质粒的表达效果,既不影响大肠杆菌生长代谢又不会造成任何无关核酸序列的残留。
为了实现本发明目的,本发明的技术方案如下:
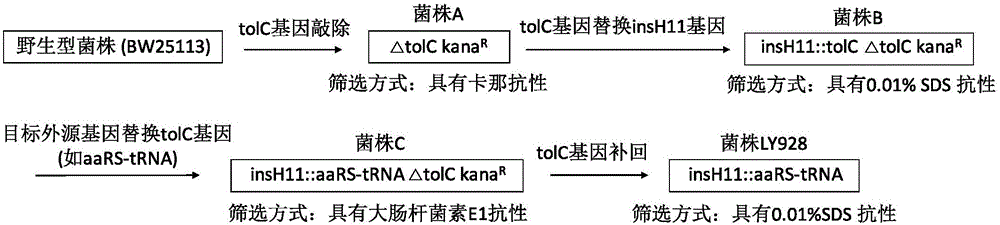
第一方面,本发明提供一种构建染色体上无痕插入外源表达系统的重组大肠杆菌的方法,包括如下步骤(重组过程均参照双质粒重组系统):
S1、将大肠杆菌菌株染色体上的tolC基因敲除,获得缺失tolC基因的菌株A;
S2、将带有大肠杆菌自身启动子序列的tolC基因重组至菌株A染色体上的insH11基因处,完全替换insH11基因序列,利用SDS进行筛选,得到菌株B;
S3、再将拟重组的外源基因重组至菌株B中,完全替换菌株B染色体上的tolC基因,利用大肠杆菌素colicin E1进行筛选,得到菌株C;
S4、将完整的野生型tolC基因重组回菌株C原有tolC基因缺失的位置,利用SDS进行筛选,即无任何无关核酸序列残留并且重组了外源基因的改造菌株。
为了保证外源基因插入的染色体位置不破坏任何已知或未知的蛋白质或RNA的功能,不给菌体带来任何的影响和改变,本发明参考大肠杆菌菌体内已有转座子插入的位置进行外源基因插入位置的选择。在进化过程中,某些DNA片段(如来自噬菌体的遗传物质)可以随机插入细菌染色体的某些位置,这些DNA片段就是转座子,最简单的转座子不含转座酶以外的任何基因序列,直接被称作插入序列(IS),它们在细菌染色体上的位置经过了漫长的自然演化与选择,能够保存在细菌染色体上的插入序列成为细菌染色体的正常组成部分,被认为对细菌正常的生长繁殖无干扰。因此本发明选择细菌染色体上的转座子位置进行替换插入。并且为了保证插入的外源基因表达不会受到抑制,可以允许高强度表达,插入位置选择距离染色体复制起始位点较近的区域。
对研究常用的大肠杆菌K12菌系已测序的MG1655菌株进行染色体插入序列的检索,发现其染色体上在不同位置有多个IS5插入序列,其中一个被标示为insH11基因,插入了染色体yhiS基因序列中,将yhiS序列分隔成yhiS1和yhiS2两个片段,并且insH11距离染色体复制起始位点较近,78.68minutes,第299,094bp-300,110bp
(gb|CP014225.1,基于大肠杆菌K12菌系菌株MG1655)。基于此位置对于K12菌系而言,天然就存在yhiS基因的插入失活,而且经过了自然界漫长的各种条件的筛选,充分验证了在这个位置外源序列的插入对菌体生长代谢的干扰可能性很小。并且此插入序列和yhiS序列在其他很多菌株中都是保守存在的(如DH10B等),因此选择将拟整合的外源基因放在此处。
利用双质粒重组系统(本发明涉及的双质粒重组系统已公开于该文献中:Gene doctoring:a method for recombineering in laboratory and pathogenic Escherichia coli strains),最终将insH11序列用拟插入的外源基因完全替换,而改造后菌株不残留任何无关核酸序列,构建过程中利用大肠杆菌的tolC蛋白进行正筛和负筛。具有完整tolC蛋白的菌株可以对抗SDS,没有tolC蛋白的菌体在SDS胁迫下无法存活。大肠杆菌素colicin E1会导致具有tolC蛋白的菌株无法存活,但缺少tolC蛋白的菌株可以在大肠杆菌素处理时存活。
所述S2具体包括如下步骤:
S21、利用引物分别扩增insH11基因及其上游272bp和下游264bp的序列,通过RF cloning插入重组质粒pDOC-H中,获得质粒pDOC-insH11;
S22、利用引物扩增带有自身启动子的tolC基因,通过RF cloning替换pDOC-insH11质粒上insH11基因的编码序列,得到重组质粒pDOC-insH11:tolC;
S23、将菌株A与S22所得重组质粒pDOC-insH11:tolC采用双质粒重组系统进行重组,利用加入0.01%SDS的培养平板进行筛选,得到菌株B。
在重组不同的外源基因时均可以使用菌株B,并且构建菌株B所使用的重组质粒pDOC-insH11:tolC通用,只需将tolC基因的序列更换成外源基因序列,重组同源臂序列不变,即可用做插入外源基因的重组质粒。
进一步地,本发明提供了利用前述方法中的S2步骤构建得到的菌株B和重组质粒pDOC-insH11:tolC,以及所述菌株B在构建无任何无关核酸序列残留的重组大肠杆菌中的应用。
进一步地,本发明还提供了大肠杆菌染色体基因组的insH11基因位点在插入外源表达系统中的应用。
所述应用具体为:将外源基因重组至大肠杆菌染色体基因组的insH11基因处,完全替换insH11基因序列,可在不影响菌体的生长和代谢的情况下,对外源基因实现高效表达,从而完全代替原始质粒的功能。
需要说明的是,某些大肠杆菌菌株的染色体上具有与所述insH11基因的序列(该序列参考已测序完整的MG1655菌株的insH11基因序列,gb|CP014225.1,第299,094bp-300,110bp)相同的序列,但该段序列可能未被明确标注,或被标注为其他名称,这些情况均不影响本发明所述方法与应用的实现,也均属于本发明的保护范围。
第二方面,本发明提供了一种可供外源基因在大肠杆菌染色体上表达的启动子和终止子序列,将可插入非天然氨基酸的正交氨酰tRNA合成酶-tRNA对基因构建在此启动子与终止子序列之间,插入染色体后此单元可以得到有效的表达,在菌体内进行非天然氨基酸的插入,其效果与在多拷贝质粒上表达的效果相当。本发明在具体实施方式中提供一种无痕的可插入非天然氨基酸p-苯甲酰苯丙氨酸(缩写为pBpa)的菌株(菌株名称为LY928)。
本发明的有益效果在于:
本发明寻找到一个普遍存在的可插入外源基因的大肠杆菌染色体位置,以及一套将质粒上的表达系统经过优化无痕插入此位置从而得到完全替代原质粒功能的重组大肠杆菌构建方法、相关重组菌株和基因重组质粒。经测试重组菌在广泛使用的培养条件下无生长和代谢障碍。并且理论分析可知,此位置进行外源基因的插入在任何条件下都不会干扰菌体的生长和代谢。
附图说明
图1本发明提供的在染色体上插入外源基因的位置。
图2为本发明提供的重组流程示意图(以实施例为例)。
图3为LY928菌株与野生型菌株BW25113生长速率的比较。
图4为LY928菌株与原质粒系统对于非天然氨基酸pBpa插入效率的比较。
具体实施方式
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。如使用不同的出发菌株,或者插入不同的外源基因,这些均属于本发明要求保护的范围。
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。
下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
实施例1LY928菌株的构建
具体步骤如下:
1.使用K12菌系的BW25113菌株作为出发菌株,将其染色体上的tolC基因进行敲除,构建菌株A。出发菌株BW25113由日本Nara科技中心(Institute of Science and Technology)免费向公众提供。
利用野生型菌株BW25113作为模板,引物tolC-5’和tolC-3’通过PCR扩增出分别带有上游324bp同源臂序列和下游280bp同源臂序列的tolC基因片段,将此片段通过RF cloning(本发明涉及的RF cloning方法已公开于该文献中:RF cloning:a restriction-free method for inserting target genes into plasmids)插入重组质粒pDOC-H中,得到质粒pDOC-tolC。利用引物tolC-kana-5’和tolC-kana-3’扩增卡那抗性基因,利用RF cloning将卡那霉素抗性基因替换pDOC-tolC质粒上tolC基因编码序列,获得敲除质粒pDOC-deltatolC:kana。所述pDOC-deltatolC:kana质粒重组所需的序列如SEQ ID NO.1~3所示。引物详见表1。
利用pDOC-deltatolC:kana质粒参考双质粒重组系统进行重组。重组子的筛选利用加入卡那霉素的培养平板,可以存活的即为阳性重组子,然后进一步测序确认重组子菌落,得到菌株A。
2.将菌株A中insH11基因用tolC基因表达单元替换,构建菌株B。
为了保证tolC蛋白的表达量可以满足重组过程中进行的正反筛选,因此仍利用原始BW25113菌株染色体上tolC基因的启动子序列,保证其表达量与野生型的一致。用此tolC表达单元完全替换insH11的表达单元(包含其启动子序列)。用野生型BW25113菌株的基因组为PCR扩增模板,利用引物isceI-insH11-up-5’和insH11-down-isceI-3’通过PCR扩增出分别带有上游272bp同源臂序列和下游264bp同源臂序列的insH11基因片段,将此片段通过RF cloning插入重组质粒pDOC-H中,得到质粒pDOC-insH11。利用引物insH11-up-tolCp-5’和Tolc-insH11-down-3’扩增带有启动子序列的tolC基因,利用RF cloning将此片段替换pDOC-insH11质粒上insH11基因编码序列,获得质粒pDOC-insH11:tolC。
所述pDOC-insH11:tolC质粒的重组所需序列如SEQ ID NO.4~6所示。引物详见表1。
利用质粒pDOC-insH11:tolC参考双质粒重组系统进行重组。重组子的筛选利用加入0.01%SDS的培养平板,可以存活的即为阳性重组子,然后进一步测序确认重组子菌落,得到菌株B。
3.修改可识别插入非天然氨基酸pBpa的氨酰tRNA合成酶-tRNA表达单元的启动子以及终止子序列,调整至合适的表达强度。
质粒在细胞内的拷贝数远多于只有一个拷贝的染色体,把在质粒上的外源表达系统原封不动的转移到染色体上,外源系统的表达量会大大降低,可能达不到所需要求的表达量。因此需要增强外源系统的表达效率,使其整合至染色体后仍可表达出可以发挥功能的量。
在研究体内蛋白质-蛋白质相互作用时,通过非天然氨基酸pBpa介导的光交联是非常有效的研究手段,而pBpa需要被在体内表达的一对正交的氨酰tRNA合成酶-tRNA对识别后才可插入目标蛋白中,而目前氨酰tRNA合成酶-tRNA对的表达依赖于质粒载体,为了使该氨酰tRNA合成酶-tRNA对整合至染色体后仍可高效率的插入pBpa,本发明对此外源系统的表达进行了优化,修改了其启动子和终止子序列。
利用辅助质粒pSup-BpaRS-6TRN(由Peter G Schultz馈赠),依据已公开的文献An enhanced system for unnatural amino acid mutagenesis in E.coi,将pSup-BpaRS-6TRN质粒上可插入非天然氨基酸pBpa的氨酰tRNA合成酶-tRNA进行优化改造,得到质粒pSuper。质粒pSuper作为PCR扩增的模板,利用引物InsH11-pSuper-5’和pSuper-insH11-down-3’将氨酰tRNA合成酶-tRNA表达单元片段扩增出来,通过RF cloning插入重组质粒pDOC-H中,得到质粒pDOC-insH11:Bpa。以质粒pDOC-insH11:Bpa为模板,通过RF cloning,利用引物tac-sd-psuper-5’,insH-tac-3’,psuper-term-3’和term-tRNA-5’修改氨酰tRNA合成酶-tRNA表达单元的启动子和终止子序列,得到重组质粒pDOC-insH11:opt-Bpa。引物详见表1,表达单元的具体序列为:
氨酰tRNA合成酶的启动子序列、氨酰tRNA合成酶基因序列和氨酰tRNA合成酶的终止子序列如SEQ ID NO.7~9所示。tRNA的启动子序列、tRNA序列和tRNA的终止子序列如SEQ ID NO.10~12所示。
需要注意的是,该启动子和终止子的组合,并不限于使用在可识别插入pBpa的氨酰tRNA合成酶-tRNA对上,其余所有的识别插入非天然氨基酸的氨酰tRNA合成酶-tRNA对均可使用此表达单元,进而重组至细菌染色体上发挥功能。
4.用可识别插入pBpa的氨酰tRNA合成酶-tRNA对表达单元替换菌株B中的tolC基因,构建上述菌株C。
利用重组质粒pDOC-insH11:opt-Bpa,在菌株B中参考双质粒重组系统进行重组,筛选重组子时加入大肠杆菌素colicin E1,可以存活的即为阳性重组子,然后进一步测序确认重组子菌落,得到菌株C。
5.将tolC基因重组回菌株C中,得到LY928菌株。
利用步骤1中所得的重组质粒pDOC-tolC,在菌株C中参考双质粒重组系统进行重组,筛选重组子时使用加入0.01%SDS的培养平板,可以存活的即为阳性重组子,然后使用引物LY928-seq-5’和LY928-seq-3’测序确认阳性重组子菌落,得到LY928菌株,引物详见表1。
表1
6.确认LY928菌株的生长速率是否受到影响
分别挑取LY928以及BW25113野生型菌株单菌落,接入LB培养基中,37度过夜培养。次日,同时1:100稀释LY928以及BW25113菌液重新接种至LB培养基中,37度培养,分别1小时,2小时,3小时,4小时,6小时,8小时和25小时这7个时间点,同时取样测量菌液在600nm处的光密度值,LY928以及BW25113均各有三组平行样品进行检测。数据见表2所示,结果显示,LY928的生长速率与BW25113菌株无区别,将insH11基因替换成修改后的氨酰tRNA合成酶-tRNA对表达单元,对菌体无影响,LY928菌株无生长缺陷。
表2
7.确认LY928菌株对于非天然氨基酸pBpa的插入效率
在蛋白翻译的过程中,氨酰tRNA合成酶-tRNA对需要识别琥珀密码子TAG,将非天然氨基酸pBpa插入目标蛋白中。为了测试LY928的插入效率,利用可表达大肠杆菌ATP合酶ε亚基的质粒载体,使用商业化的点突变试剂盒,将ε亚基的第121位氨基酸的密码子突变为琥珀密码子TAG,此质粒命名为pRoc-ε-121-TAG。将此质粒转化至LY928菌株中,同时将此质粒与pSuper质粒(可表达氨酰tRNA合成酶-tRNA对的质粒载体)共转入BW25113菌株中。分别挑取这两种转化子,接入LB培养基中,37度过夜培养。次日,同时1:100稀释至LB培养基中,每种菌液各接两份,分别一份加入终浓度100uM的pBpa,另一份不加pBpa,37度培养,长至OD600约0.6时,各取1ml菌液,使用SDS制样缓冲液制样,通过聚丙烯酰胺凝胶电泳以及免疫印迹进行检测。
结果显示,未加入pBpa时,菌体内表达出来的是分子量约16kDa的ε亚基截断蛋白(121位无法插入氨基酸而导致翻译在此位点中止)。加入pBpa时,通过菌体内表达出来的氨酰tRNA合成酶-tRNA对识别,将pBpa插入121位点,从而翻译出来分子量约18kDa的完整的ε亚基。LY928菌株插入pBpa的效率与使用pSuper质粒的效率相近,并且稍好一些。说明染色体上整合的氨酰tRNA合成酶-tRNA对可以正常表达发挥功能,LY928菌株可以完全替代质粒完成非天然氨基酸的插入。
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
- 还没有人留言评论。精彩留言会获得点赞!