利伐沙班杂质及其制备方法和用途与流程
本发明属于药物合成
技术领域:
,涉及利伐沙班相关杂质及其制备工艺。
背景技术:
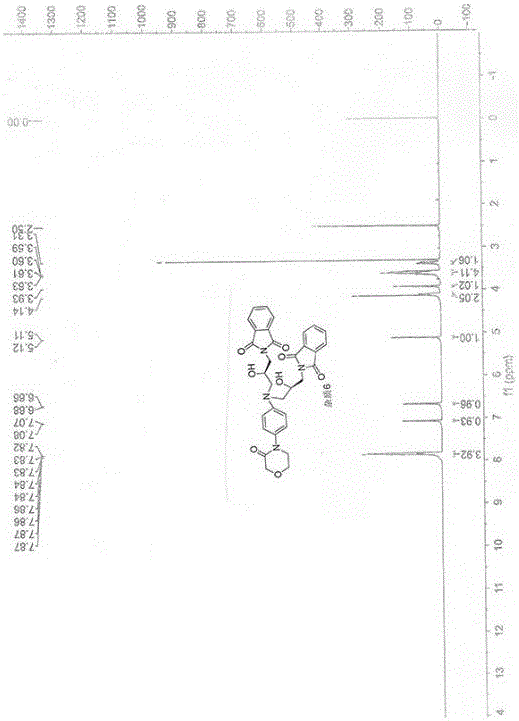
:抗凝治疗已成为目前临床预防和治疗血栓性疾病的核心和基础。其治疗药物是把“双刃剑”,抗凝效果越强,意味着出血风险就可能越大,因此,该领域药物的临床选择需特别注意风险利益比。主要有三大类药物即抗血小板聚集药物、抗凝药物和纤维蛋白溶解药组成。阿司匹林是抗血小板治疗的基础用药,疗效确切、价格低廉,是该领域的金标准。氯吡格雷与阿司匹林联合应用(双重抗血小板治疗)目前是冠心病抗血小板治疗的标准组合。但存在阿司匹林和氯吡格雷抵抗现象,且二者联用会增加出血并发症。传统抗凝药物(传统肝素、低分子肝素和华法林)因疗效确切,目前仍被广泛使用。但因其药效学或药动学不可预测、不可口服给药、受食物药物影响、剂量不固定或需监测等原因,临床应用已受到较大限制。目前,针对上述缺点已开发了两类新型抗凝药:即口服直接凝血酶抑制剂和Xa因子抑制剂。目前尚无直接证据表明,凝血酶与Xa因子哪个靶点更具优势,二者各有特点:前者作用更迅速,后者出血不良反应更小。纤维蛋白溶解药是治疗血栓的最有效方法,但目前此类上市药物存在特异性不高、半衰期短、且有内出血风险等缺点。Xa因子是外源性和内源性凝血途径的交汇点,是凝血过程中的关键点。利伐沙班是第一种口服Xa因子直接抑制剂,能高度选择性地直接抑制呈游离或结合状态的Xa因子,产生抗凝作用。利伐沙班与磺达肝素钠/肝素的本质区别在于它不需要抗凝血酶III参与,可直接拮抗游离和结合的Xa因子。而肝素则需要有抗凝血酶III才能发挥作用,且对凝血酶原复合物中的Xa因子无效。根据利伐沙班的合成工艺:利伐沙班杂质产生情况如下:杂质序号化学结构杂质1.杂质2.杂质3.杂质4.杂质5.杂质6.杂质7.杂质8.杂质9.杂质10.杂质11.杂质12.杂质13.杂质14.杂质15.目前文献J.Org.Chem.1981,46,175-177报道了杂质1、杂质15及其定向合成的方法;专利CN103896933报道了杂质2及其定向合成的方法;文献中国医药工业杂志ChineseJournalofPhannaceuticals2014,45(4)报道了杂质3、杂质4、杂质5、杂质7、杂质8、杂质9和杂质13及其定向合成的方法;专利WO2012/035057A2也报道了杂质3、杂质4和杂质5及其定向合成的方法;专利WO2004/060887和WO/2013/156936报道了杂质14的合成方法;杂质6、杂质10、杂质11和杂质12及其定向合成的方法未见相关报道。通过定向合成目标杂质,建立目标杂质的检测方法,对利伐沙班原料药的质量进行有效控制具有重要的意义。技术实现要素:本发明人首次提供了四种新物质:利伐沙班杂质6、杂质10、杂质11和杂质12及其制备方法,为利伐沙班原料药的质量进行有效控制夯实了基础。本发明的目的是提供利伐沙班的杂质化合物。本发明的另一个目的是提供上述杂质化合物的制备方法。本发明的第三个目的是提供上述杂质化合物的用途。具体地说,在本发明的实施方案中,本发明提供了四种利伐沙班杂质,即杂质6、杂质10、杂质11和杂质12,其化学结构式如下所示:第二方面,本发明提供了利伐沙班四种杂质的制备方法;其中,杂质6的制备方法,包括:a.将S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1),4-(4-氨基苯基)吗啡啉-3-酮(SM2),质子类有机溶剂混合后,搅拌加热,反应完全;b.第一次降温后过滤除去不溶物,滤液继续进行第二次降温结晶后过滤,滤饼用质子类有机溶剂重结晶。在本发明的实施方案中,本发明提供的利伐沙班杂质6的制备方法,其中,S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)用量为4-(4-氨基苯基)吗啡啉-3-酮(SM2)4~10倍(摩尔比)。在本发明的实施方案中,本发明提供的利伐沙班杂质6的制备方法,其中,加热温度为60~110℃,更优选为75~95℃。在本发明的实施方案中,本发明提供的利伐沙班杂质6的制备方法,其中,第一次降温温度为40~60℃,第二次降温温度为0~20℃。在本发明的实施方案中,本发明提供的利伐沙班杂质6的制备方法,其中,步骤a和步骤b中质子类有机溶剂为相同的,选自乙醇、甲醇、异丙醇及其醇水混合溶液。在本发明的实施方案中,本发明还提供了利伐沙班杂质10的合成路线和方法,包括:a.将2-(4-氨基苯基氨基)乙醇,S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1),质子类有机溶剂混合后,搅拌加热,反应完全;降温后过滤即得化合物I。b.将化合物I,羰基二咪唑,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后过滤,滤饼用质子类有机溶剂重结晶即得化合物II。c.将化合物II,氯乙酰甲胺,无机碱,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后过滤,滤液减压蒸干,用非质子类有机溶剂萃取,减压蒸干非质子类有机溶剂,质子类有机溶剂重结晶即得化合物III。d.将化合物III,40%甲胺水溶液,质子类有机溶剂混合后,搅拌加热,反应完全;降温酸化,再降温结晶即得化合物IV。e.将化合物IV,5-氯噻吩-2甲酸,羰基二咪唑,有机碱,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后加水结晶,过滤、干燥,再用质子类有机溶剂重结晶即得杂质10。在本发明的实施方案中,本发明提供的利伐沙班杂质10的制备方法,其中,步骤a所述S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)用量为2-(4-氨基苯基氨基)乙醇1.0~2.0倍(摩尔比);加热温度为60~90℃,更优选为75~85℃;降温温度为35~45℃;质子类有机溶剂选自乙醇、甲醇、异丙醇及其醇水混合溶液在本发明的实施方案中,本发明提供的利伐沙班杂质10的制备方法,其中,步骤b所述羰基二咪唑用量为化合物I的3.0~6.0倍(摩尔比);加热温度为50~110℃,更优选为60~90℃;降温温度为20~40℃;非质子类有机溶剂选自四氢呋喃、1,4-二氧六环、甲苯;质子类有机溶剂选自乙醇、甲醇、异丙醇及其醇水混合溶液。在本发明的实施方案中,本发明提供的利伐沙班杂质10的制备方法,其中,步骤c所述氯乙酰甲胺用量为化合物II的2.0~5.0倍(摩尔比);无机碱用量为化合物II的2.0~5.0倍(摩尔比);加热温度为50~90℃;非质子类有机溶剂选自丙酮、四氢呋喃、乙腈、甲苯;质子类有机溶剂选自乙醇、甲醇、异丙醇。在本发明的实施方案中,本发明提供的利伐沙班杂质10的制备方法,其中,步骤d所述40%甲胺用量为化合物III的8.0~15.0倍(摩尔比);加热温度为60~90℃;降温温度为0~10℃;质子类有机溶剂选自乙醇、甲醇、异丙醇。在本发明的实施方案中,本发明提供的利伐沙班杂质10的制备方法,其中,步骤e所述5-氯噻吩-2-甲酸用量为化合物IV的2.4~3.0倍(摩尔比);羰基二咪唑用量为化合物IV的3.0~4.0倍(摩尔比);有机碱用量为化合物IV的3.0~4.0倍(摩尔比);加热温度为20~40℃;非质子类有机溶剂选自N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜;质子类有机溶剂选自乙酸、乙醇、甲醇。在本发明的实施方案中,本发明还提供了利伐沙班杂质11的合成路线和方法,包括:a.将2-(2-(4-氨基苯基氨基)乙氧基)乙酸,S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1),质子类有机溶剂混合后,搅拌加热,反应完全;降温后过滤即得化合物V。b.将化合物V,羰基二咪唑,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后过滤,滤饼用质子类有机溶剂重结晶即得化合物VI。c.将化合物VI,40%甲胺水溶液,质子类有机溶剂混合后,搅拌加热,反应完全;降温酸化,再降温结晶即得化合物VII。d.将化合物VII,5-氯噻吩-2甲酸,羰基二咪唑,有机碱,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后加水结晶,过滤、干燥,再用质子类有机溶剂重结晶即得化合物VIII。e.将化合物VIII,中间体C,羰基二咪唑,有机碱,非质子类有机溶剂混合后,搅拌加热,反应完全;降温后加水结晶,过滤、干燥,再用质子类有机溶剂重结晶即得杂质11。在本发明的实施方案中,本发明提供的利伐沙班杂质11的制备方法,其中,步骤a所述S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)用量为2-(2-(4-氨基苯基氨基)乙氧基)乙酸1.0~2.0倍(摩尔比);加热温度为60~90℃,更优选为75~85℃;降温温度为35~45℃;质子类有机溶剂选自乙醇、甲醇、异丙醇及其醇水混合溶液。在本发明的实施方案中,本发明提供的利伐沙班杂质11的制备方法,其中,步骤b所述羰基二咪唑用量为化合物V的3.0~6.0倍(摩尔比);加热温度为50~110℃,更优选为60~90℃;降温温度为20~40℃;非质子类有机溶剂选自四氢呋喃、1,4-二氧六环、甲苯;质子类有机溶剂选自乙醇、甲醇、异丙醇及其醇水混合溶液。在本发明的实施方案中,本发明提供的利伐沙班杂质11的制备方法,其中,步骤c所述40%甲胺用量为化合物VI的8.0~15.0倍(摩尔比);加热温度为60~90℃;降温温度为0~10℃;质子类有机溶剂选自乙醇、甲醇、异丙醇。在本发明的实施方案中,本发明提供的利伐沙班杂质11的制备方法,其中,步骤d所述5-氯噻吩-2-甲酸用量为化合物VII的2.4~3.0倍(摩尔比);有机碱用量为化合物VII的3.0~4.0倍(摩尔比);加热温度为20~40℃;非质子类有机溶剂选自N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜;质子类有机溶剂选自乙酸、乙醇、甲醇。在本发明的实施方案中,本发明提供的利伐沙班杂质11的制备方法,其中,步骤e所述化合物VIII用量为中间体C的1.0~1.5倍(摩尔比);有机碱用量为中间体C的1.5~2.0倍(摩尔比);加热温度为20~40℃;非质子类有机溶剂选自N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、二甲亚砜;质子类有机溶剂选自乙酸、乙醇、甲醇。在本发明的实施方案中,本发明提供了利伐沙班杂质12的制备方法,包括:a.将2-((2R)-2-羟基-3-{[4-(3-氧代-4-吗啉基)苯基]氨基}丙基)-1H-异吲哚-1,3(2H)-二酮(A),40%甲胺水溶液,质子类有机溶剂混合后,搅拌加热,反应完全;b.降温后酸化,再减压蒸干溶剂,用水溶解、脱色、过滤,减压蒸干水,用非质子类有机溶剂溶解,过滤、滤液蒸干后再用非质子类有机溶剂精制。在本发明的实施方案中,本发明提供的利伐沙班杂质12的制备方法,其中,甲胺用量为2-((2R)-2-羟基-3-{[4-(3-氧代-4-吗啉基)苯基]氨基}丙基)-1H-异吲哚-1,3(2H)-二酮(A)10~15倍(摩尔比)。在本发明的实施方案中,本发明提供的利伐沙班杂质12的制备方法,其中,加热温度为60~110℃,更优选为75~85℃。在本发明的实施方案中,本发明提供的利伐沙班杂质12的制备方法,其中,降温酸化温度为0~20℃。在本发明的实施方案中,本发明提供的利伐沙班杂质12的制备方法,其中,质子类有机溶剂为选自乙醇、甲醇、异丙醇;非质子类有机溶剂选自二氯甲烷、三氯甲烷、1,2-二氯乙烷。第三方面,本发明提供了利伐沙班杂质6、利伐沙班杂质10、利伐沙班杂质11或利伐沙班杂质12作为利伐沙班中间体、原料药及其复方制剂质量研究的对照品的用途。本发明的有益结果在于:首次提供了利伐沙班杂质6、杂质10、杂质11和杂质12制备的方法;首次合成了新物质:利伐沙班杂质6、杂质10、杂质11和杂质12;且所述方法制备得到的利伐沙班杂质6的纯度在95%以上,杂质10、杂质11和杂质12的纯度均在99%以上,可以作为对照品用于的质量研究。附图说明图1.1表示的是利伐沙班杂质6的HPLC图谱。图1.2表示的是利伐沙班杂质6的氢谱。图1.3表示的是利伐沙班杂质6的质谱。图2.1表示的是利伐沙班杂质10的HPLC图谱。图2.2表示的是利伐沙班杂质10的氢谱。图2.3表示的是利伐沙班杂质10的质谱。图3.1表示的是利伐沙班杂质11的HPLC图谱。图3.2表示的是利伐沙班杂质11的氢谱。图3.3表示的是利伐沙班杂质11的质谱。图4.1表示的是利伐沙班杂质12的HPLC图谱。图4.2表示的是利伐沙班杂质12的氢谱。图4.3表示的是利伐沙班杂质12的质谱。具体实施方式下面通过本发明实施例进行具体描述本发明的实施方案。实施例1利伐沙班杂质6的制备将22.6g(111.21mmol)S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)、5.34g(27.80mmol)4-(4-氨基苯基)吗啡啉-3-酮(SM2)依次加入250ml三口瓶中,再加入无水乙醇110ml,升温回流反应24h。用自来水降温至50~55℃,抽滤,除去不溶物,收集滤液,继续降温至10~15℃析晶,过滤,得黄色固体。将此黄色固体用无水乙醇80ml回流溶解,冷却到5~10℃,过滤,滤饼于50~60℃减压干燥5小时。得产物10.0g,收率60.0%,纯度96.2%。H1-NMR(600HZ,DMSO-d6):7.86-7.87(4H,m),7.82-7.84(4H,m),7.08(2H,d),6.67(2H,d),6.67(2H,d),5.12(2H,d),4.14(2H,s),4.11(2H,m),3.93(2H,t),3.57-3.63(8H,m),3.39(2H,m);MS(m/z):599[M+H]+,621[M+Na]+实施例2利伐沙班杂质10的制备1、化合物I的制备将243.9g(1.20mol)S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)、121.8g(800.30mmol)2-(4-氨基苯基氨基)乙醇依次加入3L三口瓶中,再加入无水乙醇2.0L,升温75~85℃反应24h。用自来水降温至40~45℃,抽滤,用乙醇洗涤,收集滤饼,白色固体。于50~60℃减压干燥5小时。得化合物I的256.0g,收率90.0%。、化合物II的制备将250.0g(0.703mol)化合物I,750ml四氢呋喃加入2L三口瓶中,快速称取228.0g(1.406mol)N,N-羰基二咪唑加入反应瓶。于外温60~70℃反应10h。冷却,再加入228.0g(1.406mol)N,N-羰基二咪唑,继续于60~70℃保温回流反应10h。停止加热。降温至内温20℃,过滤,滤饼用四氢呋喃洗涤。滤饼加入2L反应瓶中,加入800ml无水乙醇,于回流下溶解。再降温至30℃;过滤,滤饼用无水乙醇洗涤。抽干,于50~60℃减压干燥5h。得化合物II227.9g,收率85.6%。、化合物III的制备将200.0g(0.524mol)化合物II,氯乙酰甲胺230.0g(2.139mol),乙腈1000ml,碳酸钾290.0g(2.101mol)加入2L三口瓶中,升温至80~90℃反应24h。降温至内温20℃,过滤,滤饼用乙腈洗涤。收集滤液,减压浓缩至干。加入2L二氯甲烷,1L水搅拌溶解萃取,分出有机层,用水洗涤,有机层用硫酸钠干燥,过滤,滤液减压浓缩至干。残留物用600ml无水乙醇重结晶,得化合物III154.1g,收率65.0%。、化合物IV的制备于3L三口瓶中,依次加入120g(0.265mol)化合物III,1.8L无水乙醇和267.0g(3.445mol)40%的甲胺水溶液,升温至75~85℃反应2~4h,降温0~10℃,滴加浓盐酸250.0g调节PH=2,控制内温0~10℃,保温搅拌结晶30~40min,过滤,用乙醇洗涤,滤饼于50~60℃减压干燥5h,得89.9g化合物IV,收率85.8%5、利伐沙班杂质10的制备将82.1g(0.505mol)5-氯-噻吩-2-甲酸(SM3),320mlN,N-二甲基甲酰胺加入1L三口瓶中搅拌溶清后,加冰浴降温至0~10℃,加入98.3g(0.606mol)N,N-羰基二咪唑,于10~20℃搅拌反应30~40min,再加入61.2g(0.606mol)三乙胺。于10~20℃搅拌反应10~15min,控制内温10~20℃,缓慢加入80.0g(0.202mol)化合物IV,升温30~35℃反应2.0~3.0h;将反应液缓慢加入装有4.0L冰水的三口瓶中,体系变为乳白色浑浊液,冰浴降温0~10℃保温搅拌30~40min。过滤,滤饼用纯化水洗涤,收集滤饼于60~70℃减压干燥12h。得到粗品。将干燥后粗品加入3.0L三口瓶,加入2L冰醋酸重结晶,得到62.0g杂质10,收率为50.2%,纯度100%。H1-NMR(600HZ,DMSO-d6):8.98(1H,t),7.63-7.70(3H,m),7.55(1H,m),7.43(2H,d),7.19(1H,d),6.93(1H,d),6.52(1H,d),4.87(1H,m),4.22(1H,t),3.90(2H,m),3.81(2H,s),3.58-3.65(4H,m),3.14(1H,m),2.59(3H,d);MS(m/z):611[M]+。实施例3利伐沙班杂质11的制备1、化合物V的制备将243.9g(0.75mol)S-(+)-N-(2,3-乙氧基丙基)邻苯二甲酰亚胺(SM1)、105.1g(0.500mol)2-(2-(4-氨基苯基氨基)乙氧基)乙酸依次加入3L三口瓶中,再加入无水乙醇1.8L,升温75~85℃反应24h。用自来水降温至40~45℃,抽滤,用乙醇洗涤,收集滤饼,白色固体。于50~60℃减压干燥5小时。得化合物V的165.4g,收率80.0%。、化合物VI的制备将150.0g(0.363mol)化合物V,400ml四氢呋喃加入2L三口瓶中,快速称取118.0g(0.726mol)N,N-羰基二咪唑加入反应瓶。于外温60~70℃反应10h。冷却,再加入118.0g(0.726mol)N,N-羰基二咪唑,继续于60~70℃保温回流反应10h。停止加热。降温至内温20℃,过滤,滤饼用四氢呋喃洗涤。滤饼加入1L反应瓶中,加入400ml无水乙醇,于回流下溶解。再降温至30℃;过滤,滤饼用无水乙醇洗涤。抽干,于50~60℃减压干燥5h。得化合物VI120.9g,收率75.8%。、化合物VII的制备将于3L三口瓶中,依次加入100.0g(0.228mol)化合物VI,1.5L无水乙醇和229.7g(2.964mol)40%的甲胺水溶液,升温至75~85℃反应2~4h,降温0~10℃,滴加浓盐酸200.0g调节PH=2,控制内温0~10℃,保温搅拌结晶30~40min,过滤,用乙醇洗涤,滤饼于50~60℃减压干燥5h,得78.4g化合物VII,收率90.0%。、化合物VIII的制备将79.7g(0.490mol)5-氯-噻吩-2-甲酸(SM3),225mlN,N-二甲基甲酰胺加入500ml三口瓶中搅拌溶清后,加冰浴降温至0~10℃,加入95.3g(0.588mol)N,N-羰基二咪唑,于10~20℃搅拌反应30~40min,再加入59.4g(0.588mol)三乙胺。于10~20℃搅拌反应10~15min,控制内温10~20℃,缓慢加入75.0g(0.196mol)化合物IV,升温30~35℃反应2.0~3.0h;将反应液缓慢加入装有3.8L冰水的三口瓶中,体系变为乳白色浑浊液,冰浴降温0~10℃保温搅拌30~40min。过滤,滤饼用纯化水洗涤,收集滤饼于60~70℃减压干燥12h。得到粗品。将干燥后粗品加入2L三口瓶,加入1.2L冰醋酸重结晶,得到化合物VIII60.4g,收率为51.5%。、利伐沙班杂质11的制备将50.0g(0.084mol)化合物VIII,100mlN,N-二甲基甲酰胺加入500ml三口瓶中搅拌溶清后,加冰浴降温至0~10℃,加入15.9g(0.098mol)N,N-羰基二咪唑,于10~20℃搅拌反应30~40min,再加入9.9g(0.098mol)三乙胺体。于10~20℃搅拌反应10~15min,控制内温10~20℃,缓慢加入22.9g(0.070mol)5-氯-N-({(5S)-2-氧代-3-[4-(3-氧代-4-吗啉基)苯基]-1,3-噁唑烷-5-基}甲胺盐酸盐(中间产品C),升温30~35℃反应2.0~3.0h;将反应液缓慢加入装有2.0L冰水的三口瓶中,体系变为乳白色浑浊液,冰浴降温0~10℃保温搅拌30~40min。过滤,滤饼用纯化水洗涤,收集滤饼于60~70℃减压干燥12h。得到粗品。将干燥后粗品加入1L三口瓶,加入500ml冰醋酸重结晶,得到29.6g杂质11,收率为48.6%,纯度99.9%。H1-NMR(600HZ,DMSO-d6):8.96(1H,m),8.05(1H,m),7.65-7.68(3H,m),7.62(2H,d),7.39(4H,t),7.17(1H,d),6.91(1H,d),6.51(1H,d),4.82(1H,m),4.75(1H,m),4.11-4.20(4H,m),3.95(2H,t),3.87(4H,m),3.78(2H,m),3.68(2H,t),3.55-3.62(4H,m),3.45(2H,m);MS(m/z):871[M]+。实施例4利伐沙班杂质12的制备于250ml三口瓶中,依次加入8.5g(21.50mmol)2-((2R)-2-羟基-3-{[4-(3-氧代-4-吗啉基)苯基]氨基}丙基)-1H-异吲哚-1,3(2H)-二酮(A),140ml无水乙醇和21.7g(280.00mmol)40%的甲胺水溶液,升温回流反应2~4h,降温10~20℃,滴加浓盐酸23.1g调节PH=2,控制内温10~15℃,保温搅拌30~40min,减压浓缩至干,用85ml纯化水溶解浓缩产物,过滤掉不溶物,滤液加入0.05g保险粉,0.5g活性炭脱色1h,过滤,滤液减压浓缩至干,加入300ml二氯甲烷溶解,过滤掉不溶物,滤液减压浓缩得白色固体7.5g,再用25ml二氯甲烷于室温下打浆0.5h,抽滤,滤饼于50~60℃减压干燥5h,得2.0g杂质12,收率75.0%,纯度99.3%。H1-NMR(600HZ,DMSO-d6):7.02(2H,t),6.59(2H,d),5.61(1H,s),4.13(2H,s),3.92(2H,m),3.60(3H,m),3.08(1H,d),2.93(1H,t),2.66(1H,m),2.53(1H,m);MS(m/z):288[M+Na]+。应用例1仪器和条件岛津高效液相色谱仪,C18柱(4.6×250mm,5μm),以缓冲盐溶液(0.01mol/L的磷酸氢二钠水溶液,用磷酸调节pH至6.0)为流动相A,以乙腈作为流动相B,检测波长为250nm,流速1.0ml/min,柱温30℃,稀释液为乙腈:水(50:50)。进样量为10μl。按如下梯度程序进行洗脱:时间(min)流动相A(%)流动相B(%)090106901025554535554535.019010459010实验步骤:取利伐沙班及各杂质适量,称重,用稀释液超声溶解,配制成每1ml中含利伐沙班和利伐沙班的杂质(杂质2、杂质3、杂质5、杂质8、杂质9、杂质10、杂质11、杂质12、杂质14、中间体B、中间体C、SM2)各约0.5μg的混合溶液。按上述条件进行高效液相分析,记录色谱图:杂质名称保留时间分离度理论塔板数拖尾因子杂质125.279——48950.941中间产品C6.1762.79252570.785SM29.6219.04183390.967杂质216.70322.290974110.784杂质321.35924.2532529161.060杂质523.0389.6722701390.734杂质824.2486.5762582861.019杂质1427.20015.3623164770.998中间产品B28.0894.5243164430.991利伐沙班29.2625.7173086571.222杂质933.65317.6562204291.031杂质1038.36113.5881426650.884杂质1138.9231.4021545541.033在上述色谱条件下,利伐沙班的杂质之间分离度好,各杂质理论板数均大于3000,且45min内结束整个有关物质检测,分析时间短,效率高,节约成本,满足中国药典的要求。应用例2仪器和条件岛津高效液相色谱仪,C18柱(4.6×250mm,5μm),以缓冲盐溶液(0.01mol/L的磷酸氢二钠水溶液,用磷酸调节pH至6.0)为流动相A,以乙腈作为流动相B,检测波长为250nm,流速1.0ml/min,柱温30℃,稀释液为乙腈:水(50:50)。进样量为10μl。按如下梯度程序进行洗脱:时间(min)流动相A(%)流动相B(%)085155851525604035604035.018515458515实验步骤:取利伐沙班及各杂质适量,称重,用稀释液超声溶解,配制成每1ml中含利伐沙班和利伐沙班的杂质(杂质2、杂质3、杂质5、杂质8、杂质9、杂质10、杂质11、杂质12、杂质14、中间体B、中间体C、SM2)各约0.5μg的混合溶液。按上述条件进行高效液相分析,记录色谱图:杂质名称保留时间分离度理论塔板数拖尾因子杂质122.447——46690.936中间产品C2.6961.73055751.094SM25.63413.25861151.092杂质28.3707.90468660.999杂质317.82631.6171348751.045杂质518.9144.333601760.966杂质820.1424.8611675761.015杂质1421.2925.6181606560.980中间产品B25.18618.2662218910.950利伐沙班26.1254.2792156081.021杂质927.6206.4872193931.063杂质1037.39831.3121480990.915杂质1138.0361.5131122061.149在上述色谱条件下,利伐沙班的杂质之间分离度好,各杂质理论板数均大于3000,拖尾因子符合要求,且45min内结束整个有关物质检测,分析时间短,效率高,节约成本,满足中国药典的要求。最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管通过参照本发明的优选实施例已经对本发明进行了描述,但本领域的普通技术人员应当理解,可以在形式上和细节上对其作为各种各样的改变,而不偏离所附权利要求所限定的本发明的精神和范围。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1