基于IonTorrent测序平台融合基因检测文库的构建方法及融合基因检测的方法与流程
本发明涉及生物技术领域,具体而言,涉及一种基于Ion Torrent测序平台融合基因检测文库的构建方法及融合基因检测的方法。
背景技术:
基因融合属于基因结构变异的一种,是指两个或多个基因的编码区首尾相连,于同一套调控序列控制之下,构成的嵌合基因。融合基因的表达产物为融合蛋白。
随着大规模平行测序(二代测序)成本的不断降低以及目标区域捕获技术的不断成熟,基于二代测序技术的基因检测得到越来越多的应用。二代测序与传统检测方法相比,具有灵敏度高,准确性高,价格低廉,可以同时检测多种靶点的优势。市面上主流的测序平台主要有两个,分别是Illumina公司的荧光可逆性终止法测序以及Thermo Fisher公司的半导体离子流测序(Ion Torrent)。
目标区域捕获利用序列捕获技术将目标区域的DNA捕捉并富集,结合高通量测序平台进行测序的研究策略。与全基因组测序相比其优势在于研究区域集中,大大降低测序费用,周期和工作量;适用于高深度测序,发现低频突变。捕获的方式主要有两种,一种是以Thermo Fisher公司为代表的基于多重PCR的扩增子捕获技术,通过在目标区域的上下游设计引物将感兴趣的区域捕获;还有就是以Agilent公司为代表的液相杂交捕获技术,该技术通过针对目标区域设计cRNA或者cDNA探针进行捕获。该方法的优势是借助探针可以捕获到目标区域的侧翼,从而可以检测与目标区域发生基因融合的区域,因此可以在DNA水平检测未知的基因融合。
Ion AmpliseqTM是Thermo Fisher公司开发的基于多重PCR对目标区域进行捕获的技术。该技术在检测融合基因方面采用的是在转录组层面(RNA水平)的扩增子建库方式,并且只能根据特定的融合位点形式设计多重PCR的引物。因此,针对每一个样本,都需要既提取DNA又提取RNA。而临床样本复杂性高,特别是福尔马林固定石蜡包埋的组织(FFPE)样本,由于保存时间较长且保存方式不规范,导致核酸的提取比较困难。并且RNA比DNA更容易发生降解,这就造成了许多样本由于RNA提取失败造成无法检测融合基因的情况发生。
技术实现要素:
本发明旨在提供一种基于Ion Torrent测序平台融合基因检测文库的构建方法及融合基因检测的方法,以解决现有技术中Ion Torrent测序平台因RNA样本不合格导致无法检测基因融合及无法检测未知融合断点的融合类型问题。
为了实现上述目的,根据本发明的一个方面,提供了一种基于Ion Torrent测序平台融合基因检测文库的构建方法。该构建方法包括以下步骤:S1,对样品DNA进行片段化,得到DNA片段;S2,对DNA片段进行末端修复及3’端添加碱基A;S3,将3’端连接有A碱基的DNA片段末端连接序列已知的接头;S4,根据序列已知的接头,进行PCR扩增,得到样本核酸文库;S5,将样本核酸文库基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因的DNA片段,采用洗脱液去除捕获过程中引入的非特异性产物;以及S6,将S5的产物通过PCR引入Ion Torrent Proton测序接头序列,得到基于Ion Torrent测序平台融合基因检测文库。
进一步地,S1中,样本DNA不足100ng时,加入500ng的载体RNA进行辅助打断。
进一步地,S1具体包括:采用超声波打断仪将样品DNA打断成主带在150bp~300bp的DNA片段。
进一步地,序列已知的接头为Illumina测序平台的测序接头。
进一步地,S6中,PCR引物的5’端含有Ion Torrent proton测序的接头序列,3’端与序列已知的接头有13bp的重叠序列。
进一步地,样品DNA来自福尔马林固定石蜡包埋的组织。
根据本发明的另一个方面,提供一种基于Ion Torrent测序平台融合基因检测的方法。该方法包括以下步骤:采用上述任一种构建方法构建基于Ion Torrent测序平台融合基因检测文库;以及在Ion Torrent的Proton测序平台上进行测序,分析测序结果得到融合基因检测的结果。
进一步地,在Ion Torrent的Proton测序平台上进行测序之前,使用安捷伦2100Bioanalyzer检测插入片段大小,以及使用qPCR定量检测文库产量。
进一步地,分析测序结果包括:过滤掉含“N”的测序序列与低质量测序序列后,采用BWA软件进行序列比对产生bam文件,再采用IGV软件进行软截断序列查看。
应用本发明的技术方案,Ion Torrent测序平台结合液相杂交捕获技术,基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因的DNA片段,然后用Ion Torrent测序平台测序,解决了现有技术中Ion Torrent测序平台因RNA样本不合格导致无法检测基因融合及无法检测未知融合断点的融合类型问题。
附图说明
构成本申请的一部分的说明书附图用来提供对本发明的进一步理解,本发明的示意性实施例及其说明用于解释本发明,并不构成对本发明的不当限定。在附图中:
图1示出了根据本发明的基于Ion Torrent测序平台融合基因检测文库的构建方法的流程示意图;
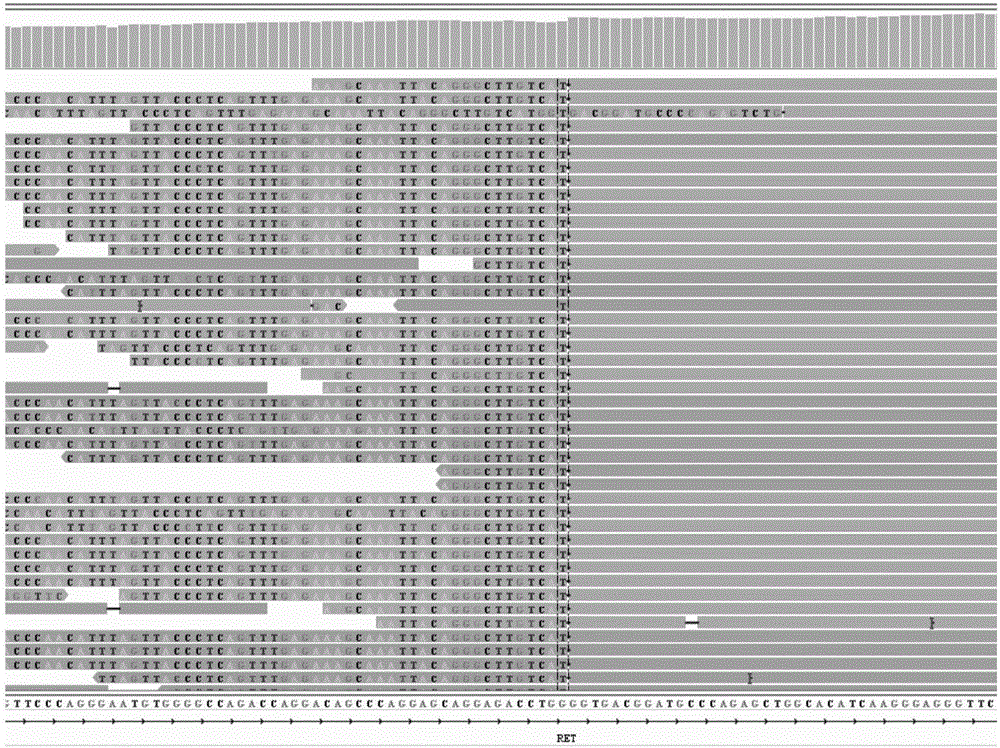
图2-1和图2-2示出了实施例1的XHC样本CCDC6-RET的融合断点IGV查看结果示意图;
图3-1和3-2示出了实施例1的DYT样本TPM3-ROS1融合断点的IGV查看结果示意图;以及
图4-1和4-2示出了实施例1的FSH样本EML4-ALK融合断点的IGV查看结果图示意图。
具体实施方式
需要说明的是,在不冲突的情况下,本申请中的实施例及实施例中的特征可以相互组合。下面将参考附图并结合实施例来详细说明本发明。
根据本发明一种典型的实施方式,提供一种基于Ion Torrent测序平台融合基因检测文库的构建方法。如图1所示,该构建方法包括以下步骤:S1,对样品DNA进行片段化,得到DNA片段;S2,对DNA片段进行末端修复及3’端添加碱基A;S3,将3’端连接有A碱基的DNA片段末端连接序列已知的接头;S4,根据序列已知的接头,进行PCR扩增,得到样本核酸文库;S5,将样本核酸文库基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因(partner gene)的DNA片段,采用洗脱液去除捕获过程中引入的非特异性产物;以及S6,将S5的产物通过PCR引入Ion Torrent Proton测序接头序列,得到基于Ion Torrent测序平台融合基因检测文库。
Ion Torrent测序平台结合液相杂交捕获技术,基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因的DNA片段,然后用Ion Torrent测序平台测序,解决了现有技术中Ion Torrent测序平台因RNA样本不合格导致无法检测基因融合及无法检测未知融合断点的融合类型问题。采用本发明的技术方案构建的目标区域捕获文库可以在Ion Torrent Proton测序仪上进行测序,能够稳定地产出数据,并且可以从微量融合阳性样本中准确检测到目标基因融合及给出正确的融合形式。
优选的,S1中,样本DNA不足100ng时,加入500ng的载体RNA(Carrier RNA)进行辅助打断。这是因为如果样本DNA不足100ng,由于DNA含量低,在超声波破碎时可能会对样本造成严重破坏,因此添加载体RNA(Carrier RNA)起到保护样本DNA的作用。
优选的,S1具体包括:采用超声波打断仪将样品DNA打断成主带在150bp~300bp的DNA片段。150bp~300bp的DNA片段适用于后续测序仪的测序读长,本发明中使用的测序仪为Ion Torrent proton,其测序读长为单端200bp,因此本步中将DNA打断成150bp-300bp长度较为合适。
根据本发明一种典型的实施方式,序列已知的接头为Illumina测序平台的测序接头。接已知序列接头的目的是为了根据已知序列设计后续PCR引物引入Ion Torrent proton测序接头。
在杂交前文库构建时添加Illumina的Y字型接头,而不是直接添加proton测序接头,存在以下优势:1)避免由于proton测序接头为平末端连接,对于微量样本而言,会由于连接效率不足导致样本DNA模板的损失,导致遗传信息的丢失;2)避免杂交使用的封闭试剂需要针对proton的接头重新设计,不能保证封闭效果和捕获效率;3)避免由于proton的测序接头中barcode序列与插入片段距离很近,无法通过PCR的手段引入barcode序列。
优选的,S6中,PCR引物的5’端含有Ion Torrent proton测序的接头序列,3’端与所述序列已知的接头有13bp的重叠序列。也就是说,在设计引物时候,引物的5’端含有Ion Torrent proton测序的接头序列,3’端含有与上一步添加的Iillumina测序接头序列互补的序列(13bp),在PCR的时候,上述的13bp长度会与Illumina测序接头序列退火复性,从而将Ion Torrent proton测序接头序列引入到PCR产物的两端。
在杂交前文库的构建过程中,添加了Illumina测序平台的测序接头,这样会导致在杂交后的PCR(引入proton测序接头)的过程中,由于引物结合的需要,需要保留一部分的Illumina接头序列,这部分序列太短的话会导致PCR失败,太长的话会导致数据浪费,经过摸索之后确定了使用13bp的overlap序列来辅助进行proton接头的引入。
根据本发明一种典型的实施方式,样品DNA来自福尔马林固定石蜡包埋的组织。
根据本发明一种典型的实施方式,提供一种基于Ion Torrent测序平台融合基因检测的方法。该方法包括以下步骤:采用上述任一种的构建方法构建基于Ion Torrent测序平台融合基因检测文库;以及在Ion Torrent的Proton测序平台上进行测序,分析测序结果得到融合基因检测的结果。
应用本发明的技术方案,Ion Torrent测序平台结合液相杂交捕获技术,基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因的DNA片段,然后用Ion Torrent测序平台测序,解决了现有技术中Ion Torrent测序平台因RNA样本不合格导致无法检测基因融合及无法检测未知融合断点的融合类型问题。
优选的,在Ion Torrent的Proton测序平台上进行测序之前,使用安捷伦2100Bioanalyzer检测插入片段大小,以及使用qPCR定量检测文库产量。
根据本发明一种典型的实施方式,分析测序结果包括:过滤掉含“N”的测序序列(reads)与低质量reads后,采用BWA软件进行序列比对产生bam文件,再采用IGV软件进行软截断reads查看。
由于Ion Torrent测序平台测序原理所限,对于串联重复碱基的测序错误率较高,在测序完成之后,仪器会自动过滤掉测序质量差的测序序列(reads),而基因融合经常会发生在串联重复碱基中,这样会导致在原始数据中会损失掉一些关键性reads。针对此问题,本发明采用查找测序最原始的电信号来找回支持融合突变的reads来保证检测的准确度。
根据本发明一种典型的实施方式,该方法包括以下步骤:
1.待检测样本DNA的片段化
使用超声波打断仪(Covaris LE220R)将样品按照表1打断参数打断成主带在150bp~300bp的DNA片段;特别地,如果样本DNA不足100ng,需加入500ng的Carrier RNA进行辅助打断。
表1
2.DNA片段末端修复及3’端添加A碱基;
3.接头的连接
将末端连接A碱基的DNA随机片段末端连接序列已知的接头(序列已知,并且在后续杂交捕获时具有针对接头序列的封闭试剂可以使用)。
4.PCR扩增
根据已知的接头序列引物,进行PCR扩增,得到样本核酸文库;
5.基于安捷伦SureSelect目标区域捕获平台的探针杂交捕获,采用洗脱液去除捕获过程中引入的非特异性产物;
6.通过PCR引入Ion Torrent Proton测序接头序列
7.文库检测
使用安捷伦2100Bioanalyzer检测插入片段大小;
使用qPCR定量检测文库产量。
下面将结合实施例进一步说明本发明的有益效果,本发明中没有详细描述的技术手段可以采用本领域的常规技术手段实现。
实施例1
非小细胞肺癌患者FFPE融合阳性样本3例,样品名称、融合基因及类型、提取量及使用量见表2。
表2
1.DNA的片段化
使用超声波破碎仪(Covaris LE220R)将目标DNA及掺入的Carrier RNA(500ng)按照特定的打断参数进行DNA的片段化,Covaris LE220R的打断参数设置如表1。
打断后的DNA经过1.8X的AMPure XP磁珠纯化后进行下一步操作。
2.末端修复及3’端加A碱基
参照表3配比准备反应混合液,用枪轻柔地上下吹吸混匀。
表3
放入PCR仪,按表4设置程序进行反应(盖温设为70℃,前30min不盖热盖,温度到达65℃,立即盖上热盖)。
表4
3.接头连接
按照表5的配比准备反应混合液,用枪轻柔地上下吹吸混匀。
表5
分为2管,每管55ul,放于PCR仪上,20℃反应15min,关闭热盖。反应结束后,使用0.8X的AMPure XP磁珠纯化DNA样本。
4.PCR扩增
按照表6的配比准备反应混合液,用枪轻柔地上下吹吸混匀。
表6
放入PCR仪,按表7设置程序反应.。
表7
用1.8X的AMPure XP磁珠对扩增产物进行纯化,最后将文库溶于30ul NF-water中。将纯化产物进行Qubit定量
5.文库杂交
5.1将文库按照上一步的浓度值,取600-700ng置于浓缩仪中蒸干至10ul,温度设置为45℃,一般需要15-20min左右。
5.2向浓缩完毕的样品中加入加入5ul的QXT快速封闭混合液以及各1ul的P5/P7接头封闭试剂(150uM),用移液器吹打混匀后短暂离心,进行下一步。
5.3后续杂交和捕获方法参照安捷伦公司SureSeclectQXT试剂盒杂交捕获方法。按照表8的反应程序杂交2h,在第三步时暂停,用以加入探针。使用DynabeadsMyone Streptavidin T1(链霉亲和素T1)磁珠进行捕获,采用洗脱液1(SureSelect Wash1)洗脱一遍,洗脱液2(SureSelect Wash2)洗脱三遍。
表8
6.捕获后文库PCR扩增引入Ion Torrent Proton测序接头序列
按照表9配置反应液,用枪轻柔地上下吹吸混匀。
表9
注:A端含有barcode的引物*中的*为代表不同barcode序列,Universal Primer P与Barcode Primer A*的序列信息见表3.
放入PCR仪,按表10设置程序进行反应。
表10
用1.8X的AMPure XP磁珠对扩增产物进行纯化,最后将文库溶于20ul NF-water中。将纯化产物进行Qubit精确定量。
表9中的引物具体信息见表10。
表10
注:X为barcode序列,对应的barcode 1-12号的序列见表11。
表11
7.文库检测
将步骤6中的纯化产物稀释到2ng/ul,取出1ul进行安捷伦2100Bioanalyzer(美国安捷伦公司)检测,构建的文库总长度在300bp左右;另外,再取出1ul用于qPCR检测,根据检测结果决定上机浓度。
根据上步所得的浓度,将文库稀释到上机要求后,在Ion Torrent的Proton测序平台上进行测序,产生400M数据。
8.下机数据分析结果
数据下机后,过滤掉含“N”的reads与低质量reads(本领域常规参数设置即可)后,采用BWA软件进行序列比对产生bam文件,再采用IGV软件进行软截断reads查看。最终确定样本是否发生了融合及融合发生的位置。表12反映了本实施例中三个样本的融合检测结果,可见本方法可以准确的检测出阳性样本中的融合基因及融合类型。图2-1到4-2为每个样本的IGV查看结果。
表12
图2-1到4-2是采用IGV软件对以上融合基因检测的图形化结果,例如图2-1和2-2展示的是XHC样本的检测结果,从图可以看出,RET基因和CCDC6两个基因都有若干序列有一部分比对不上,产生softclip序列,这些softclip序列都能比对上对方基因的参考序列。所以判断为CCDC6-RET的基因融合(图2-1到4-2用于显示序列的比对情况,而非具体的序列)。
从以上的描述中,可以看出,本发明上述的实施例实现了如下技术效果:
采用本发明提供的文库构建方法,可以准确的在DNA水平检测基因融合,这与现有的Ion Torrent建库方法相比,有以下优点:
1)应用本发明的技术方案,Ion Torrent测序平台结合液相杂交捕获技术,基于目标区域捕获平台的探针杂交捕获融合基因及伴侣基因的DNA片段,然后用Ion Torrent测序平台测序,解决了现有技术中Ion Torrent测序平台因RNA样本不合格导致无法检测基因融合及无法检测未知融合断点的融合类型问题。并且可以将单核苷酸突变(SNV)、插入缺失(indel)及拷贝数变异(CNV)共同设计进入杂交捕获的panel中,因此可以在不提取RNA的条件下同时检测四种常见的变异类型。而常规的Ion AmpliseqTM技术必须通过两个文库(DNA文库和RNA文库)才能完全检测以上四种变异类型,并且在RNA提取不成功时无法检测基因融合;
2)可以检测未知类型融合了。本方法由于采用了液相探针杂交捕获的方法进行融合基因的检测,由于探针捕获的特点,可以检测融合基因的未知融合类型。而常规的Ion AmpliseqTM技术只能根据已知的融合类型设计扩增引物,不能检测未知融合断点的基因融合;
3)本方法将液相杂交捕获技术应用在Ion Torrent测序平台,结合了Ion Torrent测序平台的测序快的特点(大约3个小时即可完成测序),大大缩短了检测的时间。
本发明针对于Ion Torrent测序平台的文库构建方法,可以在基因组层面(DNA水平)上检测驱动基因的融合以及融合基因发生的位置和形式,从而指导靶向用药治疗。检测的融合基因包括但不限于ALK,RET,ROS1等基因。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!