一种在大肠杆菌中制备基因工程IgG抗体的方法与流程
本发明属于抗体工程领域,具体涉及一种在大肠杆菌中制备基因工程IgG抗体的方法。
背景技术:
自1975年单克隆抗体技术创立以来,由于可以不受限制经过细胞杂交和筛选分泌特异性强的单克隆抗体而被大批量生产,由于该抗体分子特异性强和亲和力高的特点而深受人们的喜爱。然而,单克隆抗体也有许多问题,目前的单克隆抗体都是来自鼠源的,因而在人体治疗和使用时会产生很强的HAMA反应。这一缺陷严重的限制了单克隆抗体的应用。通过细胞融合所筛选获得的杂交瘤细胞株往往是不稳定的,很容易受到各种因素的影响而丧失抗体分泌能力,或发生所谓的返祖现象。除此以外,单克隆抗体制备技术非常费时费力。特别是在制备某些特定抗原时像小鼠这样的哺乳动物不容易产生高亲和力的抗体,从而不能对特殊抗原分子实现高灵敏度检测。因此,在体外建立一种基于大肠杆菌制备基因工程IgG抗体的方法是非常有必要的!
基因工程IgG抗体是一种新型IgG抗体类型,该抗体分子能在大肠杆菌中高效表达,其与传统的抗体分子具有相似的结构,仍保持了抗原的结合活性。然而,它的实际意义却远远超过这一点。从构建的天然噬菌体抗体库中淘选获得抗体基因,使得我们能够跨越动物免疫这一步骤,大大缩短了抗体获得的时间。另外,杂交瘤技术存在的不稳定、产量低问题,可以通过这一技术将从天然噬菌体抗体库中筛选得到的抗体基因克隆到大肠杆菌表达载体并在大肠杆菌中表达而得以克服。由于这一技术将抗体基因置于人力操纵下,抗体分子的大小、亲和力的高低、细胞毒性的强弱、抗体是否接上其他有用的分子等都可根据治疗和诊断的需要进行设计和操作。这是杂交瘤技术所不能替代的,其效率也是化学方法所不能比拟的。
相比之下,大肠杆菌表达系统受环境影响因素较少,可以比较稳定地表达抗体分子。在简单的条件下,可以实现抗体大规模的表达与纯化,从而在一定程度上提高了抗体生产的可控性,以节省大量的人力,财力和物力,实现更为可观的经济效应。同时大肠杆菌表达量远超细胞的抗体表达量,因此,该技术平台可以大大降低抗体的生产成本,以适用于不同层次、不同目的的检测需要,同时也可以作为抗体原料生产平台,为不同的用户生产定制的抗体原料。借助该平台的独特的特点,在理论上该平台可以用于针对任何抗原分子的筛选,可以不通过动物免疫筛选一些不具有免疫原性的抗原分子,从而体现了该系统具有极大的应用前景。此外,应用该技术平台可以进一步建立基于新型基因工程IgG抗体的免疫检测试剂方法。建立一套快速准确完备的对于各种食品、农产品添加剂及农药/抗生素等残留物、病原微生物以及医学领域相关的免疫检测、预警、预防科学的体系。
技术实现要素:
本发明的目的是提供一种在大肠杆菌中制备基因工程IgG抗体的方法。在大肠杆菌中高效表达纯化具有高亲和力、强特异性的基因工程IgG抗体并建立基于基因工程IgG抗体的免疫学检测方法,为最终开发基于基因工程IgG抗体的商品化免疫检测试剂盒奠定了基础。
本发明首先保护一种在大肠杆菌中制备基因工程IgG抗体的方法,通过噬菌体展示淘选和基因工程技术,分别构建抗体重链和轻链原核表达载体,然后将构建的表达载体转化到大肠杆菌中,通过IPTG诱导表达基因工程IgG抗体。
同时保护一株通过噬菌体展示淘选和基因工程技术,分别构建抗体重链和轻链原核表达载体,然后将构建的表达载体转化到大肠杆菌BL21中所获得的表达基因工程IgG抗体的大肠杆菌菌株为大肠埃希氏菌(Escherichia coli)BAC-hFLIG-Ab-001,已于2016年06月20日在中国微生物菌种保藏管理委员会普通微生物中心保藏,保藏号为CGMCC No.12641,地址为北京朝阳区北辰西路1号院。
其次保护一种在大肠杆菌中制备基因工程IgG抗体的方法所产生的基因工程IgG抗体。
所述的一种在大肠杆菌中制备基因工程IgG抗体的方法,是将噬菌体展示淘选得到的抗体重链和轻链可变区基因进行扩增,分别克隆到含有重链恒定区(CH)和轻链恒定区基因片段(CL)的原核表达载体中,并转化大肠杆菌BL21(DE3)然后进行IPTG诱导表达纯化,获得具有抗原结合活性的基因工程IgG抗体。
本发明通过在体外利用基因工程手段构建了库容量至少为109以上的天然噬菌体抗体库,利用噬菌体展示和固相淘选技术淘选获得针对特定抗原的抗体基因。将抗体的重链可变区基因VH与轻链可变区基因VL分别插入含重链恒定区CH或轻链恒定区CL的两个不同的具有质粒相容性原核表达载体中,并利用氯化钙法转化大肠杆菌。利用IPTG诱导、共培养表达重组融合蛋白,表达的抗体重链和轻链蛋白分子分别在信号肽的作用下由细胞质转运至细胞周质空间,在氧化环境中组装成完整的IgG抗体分子。组装获得的重组IgG抗体分子具有与天然IgG抗体相似的结构组成,并保留了较强的抗原识别能力和抗体特异性。利用上述方法真正实现了IgG抗体分子在细菌中的高效表达与纯化,该方法具有较强的创新性。同时,该方法所产生的基因工程IgG抗体可以应用于免疫检测试剂盒中,从而实现对食品、农产品添加剂及农药/抗生素等残留物、病原微生物以及医学领域相关的免疫检测,为商品化开发生产高质量的免疫检测试剂盒奠定了基础。
本发明的优点在于:
(1)该方法具有极强的创新性。
该方法将噬菌体展示技术、基因工程技术及大肠杆菌表达系统相结合,能够人性化的设计和筛选针对特定抗原的高亲和力的特异性IgG抗体,从而有效地解决了抗体筛选与表达难的问题。因此该抗体是比传统单克隆抗体性质优越的新型基因工程抗体。
(2)该方法解决了杂交瘤技术所存在的技术瓶颈。
从构建的天然抗体库中淘选获得抗体基因,使得我们能够跨越动物免疫这一步骤,大大缩短了抗体获得的时间。另外,杂交瘤技术存在的不稳定、产量低问题,可以通过这一技术将从天然抗体库中筛选得到的抗体基因克隆到大肠杆菌表达载体并在大肠杆菌中表达而得以克服。由于这一技术将抗体基因置于人力操纵下,抗体分子的大小、亲和力的高低、细胞毒性的强弱、抗体是否接上其他有用的分子等都可根据治疗和诊断的需要进行设计和操作。这是杂交瘤技术所不能替代的,其效率也是化学方法所不能比拟的。
(3)该方法解决了抗体在大肠杆菌中表达纯化问题。
相比其他宿主表达系统,大肠杆菌的特点是成本低,操作简单以及可以大规模培养。因此,以大肠杆菌为表达宿主来改良蛋白可溶性表达是最理想的。本方法通过多年的探索和努力,利用基因工程技术实现了抗体分子在大肠杆菌中可溶性表达、转运以及抗体分子的组装,使得我们很容易在大肠杆菌表达系统中获得高纯度的功能性基因工程IgG抗体。
(4)该平台筛选抗体分子具有高效性和可塑性。
相比传统的抗体筛选技术,该方法不需要通过动物免疫,只要获得相应浓度的抗原即可进行抗体的筛选与表达鉴定。由于库容量大,筛选获得高亲和力的特异性抗体的概率远远超过传统抗体筛选技术,可以获得多株针对特定抗原的高亲和力抗体,以满足不同检测类型的需求和抗体配对检测。此外,在获得一定亲和力的抗体株后,可以通过构建体外分子进化和基因重组技术对抗体进行人工改造,以实现不同用途和研究领域的抗体需求,从而为抗体的获得提供了保障,这一点往往是传统抗体技术所无法比拟和实现的。
(5)该平台具有广阔的市场应用价值。
大肠杆菌表达系统受环境影响因素较少,可以比较稳定地表达抗体分子。在简单的条件下,可以实现抗体大规模的表达与纯化,从而在一定程度上提高了抗体生产的可控性,以节省大量的人力,财力和物力,实现更为可观的经济效应。同时大肠杆菌表达量远超细胞的抗体表达量,因此,该技术平台可以大大降低抗体的生产成本,以适用于不同层次、不同目的的检测需要,同时也可以作为抗体原料生产平台,为不同的用户生产定制的抗体原料。借助该平台的独特的特点,在理论上该平台可以用于针对任何抗原分子的筛选,可以不通过动物免疫筛选一些不具有免疫原性的抗原分子,从而体现了该系统具有极大的应用前景。
附图说明
图1是本发明中基因工程IgG抗体分子构建示意图。
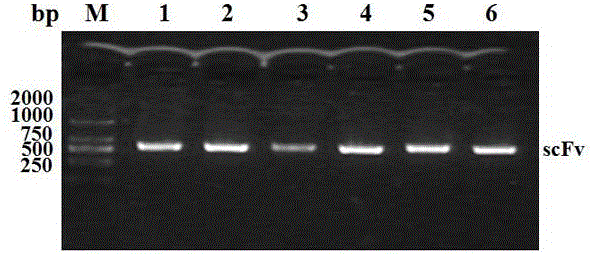
图2是本发明构建天然噬菌体抗体库PCR鉴定结果。(M:DNA marker DL-2000;泳道1-6:随机挑取单克隆子PCR扩增结果)。
图3是本发明构建天然噬菌体抗体库DNA序列分析结果。
图4是本发明基因工程IgG抗体原核表达纯化结果。(M:中分子量蛋白marker; 泳道1,3:重链和轻链空载体表达产物;泳道2,4:重链和轻链重组表达载体表达产物;泳道5-6:重链和轻链重组表达载体表达产物纯化结果)。
图5-1是本发明基因工程IgG抗体ELISA鉴定结果(实图)。
图5-2是本发明基因工程IgG抗体ELISA鉴定结果(柱状图)。
图6-1是本发明基因工程IgG抗体抗原结合活性检测结果(实图)。(1:纯化的IgG抗体HRP标记羊抗鼠IgG抗体检测结果;2:纯化的IgG抗体HRP标记抗His6标签抗体检测结果;3:阴性对照)。
图6-2是本发明基因工程IgG抗体抗原结合活性检测结果(柱状图)。
图7是本发明基因工程IgG抗体特异性检测结果。
具体实施方式
实施例1 本发明天然噬菌体抗体库的制备与鉴定。
1)、脾脏总RNA的提取
采用Trizol法进行脾脏总RNA提取。拉颈处死动物,用DEPC处理剪刀剪开小鼠腹部,迅速将脾脏取出,置于液氮预冷的研钵中加入液氮捣碎研磨成粉末。将粉末移于DEPC处理的EP管中,加入1 mL的Trizol和200 µL的氯仿,剧烈振荡混匀,10000 r/min离心5-10 min,提取上清,加入2倍体积无水乙醇或0.6倍体积的异丙醇进行RNA沉淀,离心后将沉淀吹干,加入50-100 µL的DEPC水溶解RNA。
2)、RT-PCR与抗体基因扩增
以提取的脾脏总RNA为模板,按照Promega试剂盒的步骤进行CDNA第一链的合成。利用抗体重链(引物序列为VH1BACK:5’-AGGTSMARCTGCAGSAGTCWGG-3’;VH1FOR:5’-tgaggagacggtgacggt ggtcccttggcccc-3’)和轻链的引物(引物序列为vk2back:5’-GACATTGAGCTC ACCCAGTCTCCA-3’;MJk1FONX: 5’-CCGTTTGATTTCCAGCCTGGTGCC-3’;
MJk2FONX: 5’-CCGTTTTATTTCCAGCCTGGTGCC-3’; MJk4FONX: 5’-CCG TTTTATTTCCAACCTTGTGCC-3’; MJk5FONX:5’-CCGTTTCAGCTCCAGCCT GGTGCC-3’)分别扩增VH和VL基因,用回收的VH和VL基因,与一定量的Linker DNA基因进行PCR重组,通过重叠延伸PCR扩增scFv基因。
3)、基因工程天然噬菌体抗体库的制备
PCR得到的产物经琼脂糖凝胶电泳检测后,切割凝胶上的目的条带经Fermentas公司胶回收试剂盒进行回收目的DNA片段。将回收得到的PCR产物经Sfi I和 Not I双酶切后与同样酶切的载体pCANTAB-5E DNA进行连接,二者的连接反应摩尔比例为3:1。16℃进行连接过夜。将过夜连接的DNA通过电击转化100 µL大肠杆菌TG1感受态细胞中,立即加入10 mL 2×YTG(含有2%的葡萄糖)培养基,37℃培养,待菌体生长至OD600为0.8时,加入M13KO7辅助噬菌体,至终浓度1010 pfu/mL。30 min后再加入Amp抗生素至终浓度100 μg/mL,继续培养1 h,1000 g,4℃离心收集菌体沉淀;向沉淀中加入新鲜的YT培养基(含有100 μg/mL Amp和50 μg/mL Kana),37℃培养过夜;低温下,1000 g离心收集上清,利用PEG/NaCl沉淀噬菌体(体积比为上清:PEG/NaCl=5 :1),用新鲜的YT培养基溶解噬菌体,所得的溶液为噬菌体抗体初级库。使用不同梯度稀释的噬菌体,各取10 µL涂布于表层琼脂糖的LB平板,计算库容量。
4)、基因工程天然噬菌体抗体库的鉴定
随机挑选20个SOB-AG平板上的细菌单菌落转化子,接种与4 mL 2×YTG(含有2%的葡萄糖)培养基,37℃培养过夜,利用PCR对20个不同的转化子进行PCR扩增鉴定。同时,利用噬菌体载体通用引物(pCANTAB5-R1: 5’-CCATGATTACGCCAAGCTTTGGAGCC-3’ )对所挑取的转化子进行DNA测序分析。
抗体基因的淘选
1)、单链抗体的淘选
用pH为9.6的蛋白包被液将抗原稀释成浓度为5 µg/mL, 进行96孔板包被,100 µL/孔,于4℃环境中包被过夜。用移液器吸走蛋白包被液,PBS洗3次,以除去游离的抗原分子,向孔中加入200 µL/孔的PBSM封闭液,置于37℃封闭2 h。弃去封闭液,向孔内加入已经制备好的噬菌体抗体库,100 µL/孔,置于37℃反应2 h。分别用PBST和PBS洗孔10次,200 µL/孔。向反应孔中加入Gly-HCL,利用该溶液的酸性条件洗脱与抗原结合的噬菌体,再用1 mol/L Tris-HCL(pH 8.0)中和Gly-HCL。立即取出上述洗脱中和的噬菌体进行TG1细胞侵染,以扩增噬菌体数目便于下一轮的淘选。照此方法进行连续6轮的富集淘选,从第三轮开始,每轮淘选得到的噬菌体各取10 µL进行涂布。
2)、抗原特异性单链抗体的ELISA检测
第三、四、五、六轮的涂布的SOB-AG平板上各挑取25个单链抗体的单菌落,按照上述的方法进行噬菌体抗体的制备。用pH为9.6的蛋白包被液将抗原稀释成浓度为5 µg/mL, 进行96孔板包被,100 µL/孔,于4℃环境中包被过夜。用移液器吸走蛋白包被液,PBS洗3次,以除去游离的抗原分子,向孔中加入200 µL/孔的PBSM封闭液,置于37℃封闭2 h。弃去封闭液,向孔内分别加入上述单菌落的噬菌体抗体,100 µL/孔,做好记录,置于37℃反应2 h。分别用PBST和PBS洗孔10次,200 µL/孔。加入PBSM稀释的抗pⅢ蛋白的单克隆抗体(1:4000稀释),置于37℃反应2 h。 重复步骤6一次,加入PBSM稀释的HRP标记的羊抗鼠IgG抗体(1:8000稀释),置于37℃反应2 h。再重复步骤6一次。每孔加入100 µL的TMB显色缓冲液,37℃反应15 min,加入2M H2SO4终止反应。OD450 nm测定吸收值。所有实验进行3个平行,以M13KO7设为阳性,而BSA设为阴性对照。
3)、抗原特异性单链抗体的鉴定
所有阳性的单链抗体株重组质粒的DNA测序均在华大基因公司进行,测序后的序列直接与NCBI数据库进行比对分析,测序时采用的引物序列为:pCANTAB5-R1: 5’-CCA TGA TTA CGC CAA GCT TTG GAG CC-3’。通过DNA测序获得抗体重链和轻链可变区基因序列。
基因工程IgG抗体原核表达载体的构建与表达纯化
利用EcoRI和Hind III酶切位点将抗体VH基因插入pET-his6-sp-ch-his6载体中,形成重组载体pET-his6-sp-vh-ch-his6,将测序正确的重组质粒在大肠杆菌BL21(DE3)中进行融合蛋白表达(如图1 所示)。表达的蛋白含有His6 tag标签。利用Sph I和Cla I酶切位点将抗体VL基因插入pACYC-sp-cl-c-myc载体中,形成重组载体pACYC-sp-vl-cl-c-myc,将测序正确的重组质粒在大肠杆菌BAC-hFLIG-Ab-001中进行融合蛋白表达。表达的蛋白含有c-myc标签(如图1 所示)。蛋白SDS-PAGE分析结果如图4所示:与对照菌株和空载体对照相比,分别在55 kDa和25 kDa处有两条明显的融合蛋白表达条带,纯化的蛋白条带也分别位于55kDa和25 kDa处。由于表达的IgG抗体分子的重链含有His-6 tag标签,而轻链片段则不含有His-6 tag标签。本研究利用镍离子亲和层析和抗原抗体相互作用组装原理进行IgG抗体纯化,最终获得完整的IgG抗体分子。SDS-PAGE分析结果有重链和轻链两条蛋白表达条带,表明,原核表达载体的构建是成功的,抗体的重链和轻链片段能够发生相互作用并组装成IgG抗体,可以进行后续的相关实验。
大肠杆菌中表达基因工程IgG抗体的ELISA鉴定
将纯化的基因工程IgG抗体(1:50; 1:100;1:200稀释)和BSA抗原用包被缓冲液稀释至5.0 μg/mL的浓度,分别包被96孔酶标板,100 μL/孔,漂洗后并用4%的PBSM封闭,200 μL/孔。用5% PBSM 1:8000分别稀释HRP标记的抗6×His抗体和HRP标记的羊抗人IgG抗体,37℃孵育1.5 h,重复洗涤步骤,加入TMB显色液100 μL/孔,37℃孵育10~15 min,并加入2 mol/L H2SO4终止液。用酶标仪测450 nm处的OD值。如图5 所示,ELISA结果显示,表达的抗体的重链和轻链发生组装并被利用亲和层析方法成功纯化出来,纯化的基因工程IgG抗体能够被HRP标记的抗6×His抗体和HRP标记的羊抗人IgG抗体分别识别,说明通过该方法表达和纯化基因工程IgG抗体是成功的。
大肠杆菌中表达基因工程IgG抗体的活性测定
用包被缓冲液(pH 9.6)将靶抗原(TLH)稀释至2.5 μg/mL的浓度,洗涤封闭如同前面ELISA方式进行,分别加入不同稀释浓度的纯化的基因工程IgG抗体,用HRP标记羊抗人IgG抗体间接检测IgG抗体与抗原的结合活性,以OD450 nm值分析抗体的特异性。如图6所示,该IgG抗体能够和靶抗原发生结合,且与加入抗体的浓度呈正相关性,同时,该抗体而不与阴性抗原发生结合。ELISA结果表明,该方法高效表达具有抗原结合活性的IgG抗体是可行的。
大肠杆菌中表达基因工程IgG抗体的特异性测定
用包被缓冲液(pH 9.6)将不同的抗原(1-2: TLH、阴性:BSA、Control 1: OVA、Control 2: KLH)稀释至2.5 μg/mL的浓度,洗涤封闭如同前面ELISA方式进行,抗TLH的抗血清用于做阳性对照,以OD450 nm值分析抗体的特异性。如图7所示,该IgG抗体只特异性识别TLH抗原,而与其他相关的抗原分子不发生反应。表明该基因工程IgG抗体特异性较好,与其他抗原几乎没有交叉反应。
以上所述仅为本发明的较佳实施例,凡依本发明申请专利范围所做的均等变化与修饰,皆应属本发明的涵盖范围。
SEQUENCE LISTING
<110> 福州康和尔生物科技有限公司
<120> 一种在大肠杆菌中制备基因工程IgG抗体的方法
<130> 9
<160> 9
<170> PatentIn version 3.3
<210> 1
<211> 22
<212> DNA
<213> 人工序列
<400> 1
aggtsmarct gcagsagtcw gg 22
<210> 2
<211> 32
<212> DNA
<213> 人工序列
<400> 2
tgaggagacg gtgacggtgg tcccttggcc cc 32
<210> 3
<211> 24
<212> DNA
<213> 人工序列
<400> 3
gacattgagc tcacccagtc tcca 24
<210> 4
<211> 24
<212> DNA
<213> 人工序列
<400> 4
ccgtttgatt tccagcctgg tgcc 24
<210> 5
<211> 24
<212> DNA
<213> 人工序列
<400> 5
ccgttttatt tccagcctgg tgcc 24
<210> 6
<211> 24
<212> DNA
<213> 人工序列
<400> 6
ccgttttatt tccaaccttg tgcc 24
<210> 7
<211> 24
<212> DNA
<213> 人工序列
<400> 7
ccgtttcagc tccagcctgg tgcc 24
<210> 8
<211> 26
<212> DNA
<213> 人工序列
<400> 8
ccatgattac gccaagcttt ggagcc 26
<210> 9
<211> 26
<212> DNA
<213> 人工序列
<400> 9
ccatgattac gccaagcttt ggagcc 26
- 还没有人留言评论。精彩留言会获得点赞!