聚酰亚胺前驱物组合物、聚酰亚胺的制备方法及聚酰亚胺与流程
本发明是有关一种聚酰亚胺前驱物组合物及其应用,特别是提供一种可制得具有良好机械性质与薄膜性质的聚酰亚胺的聚酰亚胺前驱物组合物。
背景技术:
聚酰亚胺具有耐热氧化性、耐热性、耐放射性及耐化性等优点,故聚酰亚胺常被应用于汽车材料、航空材料、绝缘材料及液晶配向膜等应用领域。聚酰亚胺一般是通过对聚酰胺酸进行环化反应(即酰亚胺化反应)制得。其中,聚酰胺酸是利用二酐化合物与二胺化合物经反应所形成。
然而,基于二胺化合物的总使用量为100摩尔百分比,当二酐化合物的总使用量不小于98摩尔百分比时,所形成的聚酰胺酸链段将会无限延伸,而增加所制得聚酰胺酸的分子量,进而大幅提升其粘度。因此,当聚酰胺酸利用涂布成膜的方式制作聚酰亚胺时,过高的粘度会降低聚酰胺酸的涂布性,而难以涂布成膜。
为了解决前述粘度过大所造成的涂布缺陷,通过调整前述反应的反应条件,聚酰胺酸的分子量可被降低,而具有适当的粘度,并可涂布成膜,进而可经环化反应制得聚酰亚胺膜。但是,当聚酰胺酸的分子量降低时,所制得聚酰亚胺膜的机械性质亦随之降低,而无法满足应用的需求。
此外,于前述的反应中,为了获得适当的涂布粘度,其需借由繁复实验方可获得适当的参数条件,而需耗费大量的时间成本,并徒增制备成本。再者,所制得的聚酰胺酸仍无法兼顾涂布性与机械性质。
有鉴于此,急需提供一种聚酰亚胺前驱物组合物及其应用,以改进已知聚酰亚胺前驱物组合物及其应用的缺陷。
技术实现要素:
因此,本发明的一个方面在于提供一种聚酰亚胺前驱物组合物,其通过使单体化合物与聚酰胺酸产生反应,以制得具有良好机械性质与薄膜性质的聚酰亚胺。
本发明的另一个方面在于提供一种聚酰亚胺的制备方法,其是利用前述的聚酰亚胺前驱物组合物进行反应所形成。
本发明的又一个方面在于提供一种聚酰亚胺,其是通过前述的制备方法所制得。
本发明的再一个方面在提供一种聚酰亚胺,此聚酰亚胺是由前述的制备方法所制得,且具有特定的重复单元。
根据本发明的一个方面,提出一种聚酰亚胺前驱物组合物。此聚酰亚胺前驱物组合物包含聚酰胺酸及单体化合物,其中聚酰胺酸是由单体混合物经第一反应所制得,且单体混合物包含二胺化合物及二酐化合物。此单体化合物具有两个羧基及两个酯基。
依据本发明的一实施例,前述的单体化合物具有如下式(i)所示的结构:
于式(i)中,r1代表
依据本发明的另一实施例,基于前述二胺化合物的总使用量为100摩尔百分比,单体化合物的使用量为1摩尔百分比至10摩尔百分比。
根据本发明的另一个方面,提出一种聚酰亚胺的制备方法。此方法是先提供单体混合物,并对此单体混合物进行第一反应,以制得聚酰胺酸。其中,单体混合物包含二胺化合物及二酐化合物。
然后,混合单体化合物及聚酰胺酸,以形成聚酰亚胺前驱物,其中单体化合物具有两个羧基及两个酯基。接着,对聚酰亚胺前驱物进行第二反应,以获得聚酰亚胺。
依据本发明的一实施例,前述的二酐化合物包含苯均四羧酸二酐、3,3’,4,4’-联苯四羧酸二酐、3,3’,4,4’-二苯甲酮四羧酸二酐、4,4’-氧二磷苯二酸二酐、3,3’,4,4’-二苯基砜四羧酸二酐、乙二醇-双偏苯三酸酐或双酚a型二醚二酐。
依据本发明的另一实施例,前述的二胺化合物包含对苯二胺或9,9’-双(4-氨基苯基)芴。
依据本发明的又一实施例,前述的单体化合物具有如下式(i)所示的结构:
于式(i)中,r1代表
依据本发明的再一实施例,基于前述二胺化合物的总使用量为100摩尔百分比,二酐化合物的使用量大于或等于90摩尔百分比且小于98摩尔百分比,且单体化合物的使用量为1摩尔百分比至10摩尔百分比。
根据本发明的又一个方面,提出一种聚酰亚胺。此聚酰亚胺是通过前述的方法反应单体混合物及单体化合物而制得。其中,单体混合物包含二胺化合物及二酐化合物。
根据本发明的又一个方面,提出一种聚酰亚胺。此聚酰亚胺具有如下式(ii-1)与式(ii-2)所示的重复单元:
于式(ii-1)中,ar1代表
于式(ii-2)中,ar2代表衍生自该二酐化合物的二价有机基团,且a代表衍生自该二胺化合物的二价有机基团。
本发明的聚酰亚胺前驱物组合物及其应用,首先,借由使聚酰胺酸与单体化合物产生反应,以延伸所制得聚酰亚胺的分子链长度,而可使其具有良好的机械性质与薄膜性质。其次,通过单体化合物的导入,所制得的聚酰亚胺具有适当的分子量,从而可抑制因分子链延长所造成的粘度上升,进而易于加工。
附图说明
图1是绘示依照本发明的一实施例的聚酰亚胺的制备方法的流程图;
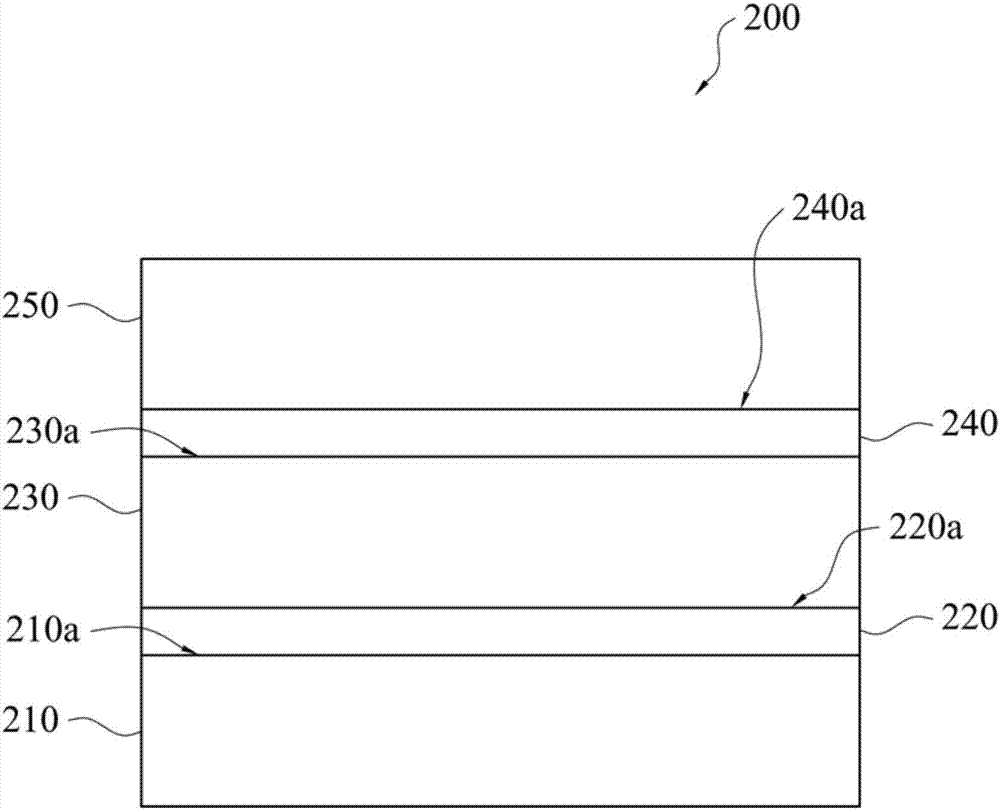
图2是绘示依照本发明的一实施例的聚酰亚胺积层板的侧视图。
其附图标记为:
100:方法
110:提供单体混合物的步骤
120:对单体混合物进行第一反应,以获得聚酰胺酸的步骤
130:混合单体化合物及聚酰胺酸,以形成聚酰亚胺前驱物的步骤
140:对聚酰亚胺前驱物进行第二反应的步骤
150:获得聚酰亚胺的步骤
200:聚酰亚胺积层板
210:第一铜箔
220:第一热塑性聚酰亚胺层
230:聚酰亚胺层
240:第二热塑性聚酰亚胺层
250:第二铜箔
210a/220a/230a/240a:侧面
具体实施方式
以下仔细讨论本发明实施例的制备和使用。然而,可以理解的是,实施例提供许多可应用的发明概念,其可实施于各式各样的特定内容中。所讨论的特定实施例仅供说明,并非用以限定本发明的范围。
请参照图1,其是绘示依照本发明的一实施例的聚酰亚胺的制备方法的流程图。在一实施例中,方法100是先提供单体混合物,并对单体混合物进行第一反应,以获得聚酰胺酸,如步骤110及120所示。
前述的单体混合物可包含二胺化合物及二酐化合物。此二胺化合物可包含但不限于3,4’-二氨基二苯基醚(3,4’-diaminodiphenylether)、4,4’-二氨基二苯基醚(4,4’-diaminodiphenylether)、对苯二胺(p-phenylenediamine)、间苯二胺(m-phenylenediamine)、3,5-二氨基苯甲酸(3,5-diaminobenzoicacid)、2,2’-双(4-氨基苯基)丙烷[2,2’-bis(4-aminophenyl)propane]、4,4’-二氨基二苯基甲烷(4,4’-diaminodiphenylmethane)、4,4’-二氨基二苯基砜(4,4’-diaminodiphenylsulfone)、3,3’-二氨基二苯基砜(3,4’-diaminodiphenylsulfone)、4,4’-二氨基二苯基硫醚(4,4’-diaminodiphenylsulfide)、1,3-双(4-氨基苯氧基)苯[1,3-bis(4-aminophenoxy)benzene]、1,3-双(3-氨基苯氧基)苯[1,3-bis(3-aminophenoxy)benzene]、1,4-双(4-氨基苯氧基)苯[1,4-bis(4-aminophenoxy)benzene]、4,4-双(4-氨基苯氧基)-联苯[4,4-bis(3-aminophenoxy)biphenyl]、2,2’-双[4-(4-氨基苯氧基)苯基]丙烷{2,2’-bis[4-(4-aminophenoxy)phenyl]propane}、2,2’-双[4-(3-氨基苯氧基苯)基]丙烷{2,2’-bis[4-(3-aminophenoxy)pheny]propane}、2,2’-二甲基-4,4’-二氨基联苯(2,2’-dimethyl-4,4’-diaminobiphenyl)、3,3’-二甲基-4,4’-二氨基联苯(3,3’-dimethyl-4,4’-diaminobiphenyl)、3,3’-二烃基-4,4’-二氨基联苯(3,3’-dihydroxybiphenyl-4,4’-diamino)、9,9’-双(4-氨基苯基)芴[9,9’-bis(4-aminophenyl)fluorene]、2,2’-双(4-[3-氨基苯氧基]苯基)砜[2,2’-bis(4-(3-aminophenoxy)phenyl)sulfon]、2,6-二氨基嘧啶(2,6-diaminopyridine)、聚丙烯醚二胺(polyoxypropylenediamine)、4,4’-(1,3-二异丙烷基苯)二苯胺[4,4’-(1,3-phenylenediisopropylidene)bisaniline;bisaniline-m]、4,4’-(1,4-二异丙烷基苯)二苯胺[4,4’-(1,4-phenylenediisopropylidene)bisaniline;bisaniline-p]、冰片烷二胺(norbornanedimethylamine;nbda)、其他适当的二胺化合物或上述二胺化合物的任意混合。
二酐化合物可包含但不限于苯均四羧酸二酐(pyromelliticdianhydride)、3,3’,4,4’-联苯四羧酸二酐(3,3’,4,4’-biphenyltetracarboxylicdianhydride)、3,3’,4,4’-二苯甲酮四羧酸二酐(3,3’,4,4’-benzophenonetetracarboxylicdianhydride)、4,4’-氧二邻苯二酸二酐(4,4’-oxydiphthalicdianhydride)、3,3’,4,4’-二苯基砜四羧酸二酐(3,3’,4,4’-diphenylsulfonetetracarboxylicdianhydride)、乙二醇-双偏苯三酸酐(ethyleneglycol-bis-trimellitateanhydride)、双酚a型二醚二酐{2,2-bis[4-(3,4-dicarboxyphenoxy)phenyl]propanedianhydride}、其他适当的二酐化合物或上述二酐化合物的任意混合。
基于二胺化合物的总使用量为100摩尔百分比,此二酐化合物的使用量可为大于或等于90摩尔百分比且小于98摩尔百分比,较佳为大于或等于94摩尔百分比且小于98摩尔百分比,且更佳可为96摩尔百分比且97摩尔百分比(即大于或等于96摩尔百分比且小于或等于97摩尔百分比)。
若二酐化合物的使用量不小于98摩尔百分比时,所制得聚酰胺酸的分子链长度易增加,而徒增其分子量,进而增加聚酰胺酸的粘度,并易形成半固态的凝胶状,因此无法涂布成膜。
若二酐化合物的使用量小于95摩尔百分比,虽然所制得的聚酰胺酸的末端基可为二胺基团,只有聚酰胺酸的分子量较低,当进行后续的第二反应后,所形成的聚酰亚胺具有较低的粘度,且其加工性较差,而难以有效满足应用的需求。
当二酐化合物的使用量为前述的范围时,所制得的聚酰胺酸可具有适当的分子链,而具有适当的分子量,进而具有适当的粘度,因此有助于涂布形成后续的聚酰亚胺层。
在一实施例中,所制得聚酰胺酸的分子量可为50000至150000。依据应用的需求,此聚酰胺酸的分子量可适当地调整,只有此聚酰胺酸须可进一步与后续的单体化合物进行第二反应,并形成聚酰亚胺或聚酰亚胺层。
在进行步骤120后,混合一单体化合物及前述的聚酰胺酸,即可形成本发明的聚酰亚胺前驱物,如步骤130所示。
前述的单体化合物具有两个羧基及两个酯基。在一实施例中,单体化合物可具有如下式(i)所示的结构:
于式(i)中,r1代表
前述的单体化合物可单独一种或混合多种使用。
前述的r1较佳可为
请继续参照图1。于进行步骤130后,对聚酰亚胺述的前驱物进行第二反应,即可获得本发明的聚酰亚胺,如步骤140及150所示。
于步骤130及140中,单体化合物可通过羧基与酯基分别与两个聚酰胺酸链段的末端基团(即胺基团)进行第二反应,而可延长所制得聚酰亚胺的分子链,进而使具有较低分子量的聚酰胺酸反应形成具有较高分子量的聚酰亚胺,因此可提升所制得聚酰亚胺的机械性质。
据此,通过单体化合物的导入,本发明可有效克服已知聚酰亚胺无法兼顾机械性质及薄膜性质的缺陷。
因此,基于前述二胺化合物的总使用量为100摩尔百分比,单体化合物的使用量可为1摩尔百分比至10摩尔百分比,较佳为1摩尔百分比至5摩尔百分比,且更佳为2摩尔百分比至3摩尔百分比。
若单体化合物的使用量小于1摩尔百分比时,过少的单体化合物限制聚酰胺酸链段的结合,而缩短所制得聚酰亚胺的分子链长度,进而降低其成膜性,因此具有较差的薄膜性质。
在一实施例中,于步骤140及150中,前述所制得的聚酰亚胺可具有如下式(ii-1)与式(ii-2)所示的重复单元(repeatingunit):
于式(ii-1)中,ar1可代表衍生自具有两个羧基及两个酯基的单体化合物的二价有机基团,且a可代表衍生自前述二胺化合物的二价有机基团;以及
于式(ii-2)中,ar2可代表衍生自前述二酐化合物的二价有机基团,且a可代表衍生自前述二胺化合物的二价有机基团。
在一实施例中,前述聚酰亚胺可由式(ii-1)及式(ii-2)所示的重复单元所组成,其中此聚酰亚胺可为嵌段共聚物(blockcopolymer)或随机共聚物(randomcopolymer)。
基于前述所使用的单体化合物的不同,ar1可具有不同的结构。
在一实施例中,ar1可代表
在另一实施例中,前述的ar1较佳可为
其中,基于聚酰亚胺为100%,如式(ii-1)所示的重复单元于所制得聚酰亚胺的含量可为1%至10%,较佳为1%至5%,且更佳可为2%至3%。
在一具体例中,本发明所制得的聚酰亚胺积层板的挠曲性(flexuralendurance)可大于10000。
聚酰亚胺积层板的制备方法
聚酰亚胺积层板的制备方法为本案所属技术领域具有通常知识者所熟知。因此,以下仅简单地进行陈述。
请参照图2,其是绘示依照本发明的一实施例的聚酰亚胺积层板的侧视图。此聚酰亚胺积层板200包含第一铜箔210、设于第一铜箔210上的第一热塑性聚酰亚胺层220、设于第一热塑性聚酰亚胺层220上的聚酰亚胺层230、设于聚酰亚胺层230上的第二热塑性聚酰亚胺层240,以及设于第二热塑性聚酰亚胺层240上的第二铜箔250。
其中,于聚酰亚胺积层板200的制备方法中,热塑性聚酰胺酸溶液先涂布于该第一铜箔210的侧面210a上,并以80℃至190℃的温度去除溶剂,而可形成第一热塑性聚酰胺酸层。
然后,将本发明所制得的聚酰亚胺前驱物组合物涂布于第一热塑性聚酰胺酸层的侧面220a上。加热至80℃至190℃以去除溶剂,而可形成聚酰胺酸层。
相同地,将第二热塑性聚酰胺酸涂布于前述聚酰胺酸层的侧面230a上,并加热以去除溶剂,即可形成第二热塑性聚酰胺酸层。
接着,对积层有第一铜箔、第一热塑性聚酰胺酸层、聚酰胺酸层及第二热塑性聚酰胺酸层的积层板进行环化反应(即酰亚胺化反应),即可制得具有第一热塑性聚酰亚胺层220、聚酰亚胺层230及第二热塑性聚酰亚胺层240的积层板。
之后,将第二铜箔250设于第二热塑性聚酰亚胺层240的侧面240a上,并进行压合制程,即可制得本发明的聚酰亚胺积层板200。其中,压合工艺的温度可为320℃至380℃,且其压力可为50kgf/cm2至100kgf/cm2。
以下利用实施例以说明本发明的应用,然其并非用以限定本发明,任何熟习此技艺者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰。
制备聚酰胺酸
合成例1
在通入氮气的1000毫升的四口反应器中,加入19.630克(0.182摩尔)的对苯二胺、2.639克(0.0075摩尔)的9,9’-双(4-氨基苯基)芴及425克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将7.434克(0.034摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将44.565克(0.151摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得合成例1的聚酰胺酸。
合成例2
在通入氮气的1000毫升的四口反应器中,加入26.112克(0.242摩尔)的对苯二胺、3.511克(0.01摩尔)的9,9’-双(4-氨基苯基)芴及400克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将9.888克(0.045摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将58.54克(0.199摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得合成例2的聚酰胺酸。
合成例3
在通入氮气的1000毫升的四口反应器中,加入32.491克(0.3摩尔)的对苯二胺、4.368克(0.013摩尔)的9,9’-双(4-氨基苯基)芴及375克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将12.304克(0.056摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将70.995克(0.241摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得合成例3的聚酰胺酸。
制备聚酰亚胺前驱物组合物
以下是根据表1制备实施例1至实施例3及比较例1至比较例3的聚酰亚胺前驱物组合物。其中,为了比较的方便性,实施例1至实施例3所对应使用的合成例1至合成例3的二酐化合物与二胺化合物的使用量是一并表列于表1中。
实施例1
实施例1的聚酰亚胺前驱物组合物是将0.731克(约0.002摩尔)的3,3’,4,4’-二苯基二羧酸二乙酯加至合成例1的聚酰胺酸中,混合均匀后,即可制得实施例1的聚酰亚胺前驱物组合物。其粘度约为12000cps,且固含量为15%。所制得的聚酰亚胺前驱物组合物以下述涂布性及薄膜性质的评价方式进行评价,所得结果如表1所示。
实施例2
实施例2的聚酰亚胺前驱物组合物是将1.945克(约0.005摩尔)的3,3’,4,4’-二苯基二羧酸二乙酯加至合成例2的聚酰胺酸中,混合均匀后,即可制得实施例2的聚酰亚胺前驱物组合物。其粘度约为12000cps,且固含量为20%。所制得的聚酰亚胺前驱物组合物以下述涂布性及薄膜性质的评价方式进行评价,所得结果如表1所示。
实施例3
实施例3的聚酰亚胺前驱物组合物是将4.841克(约0.013摩尔)的3,3’,4,4’-二苯基二羧酸二乙酯加至合成例3的聚酰胺酸中,混合均匀后,即可制得实施例3的聚酰亚胺前驱物组合物。其粘度约为12000cps,且固含量为25%。所制得的聚酰亚胺前驱物组合物以下述涂布性及薄膜性质的评价方式进行评价,所得结果如表1所示。
比较例1
在通入氮气的1000毫升的四口反应器中,加入19.675克(0.182摩尔)的对苯二胺、2.645克(0.008摩尔)的9,9’-双(4-氨基苯基)芴及425克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将7.451克(0.034摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将45.225克(0.154摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得比较例1的聚酰亚胺前驱物组合物,但此聚酰亚胺前驱物组合物(即聚酰胺酸)为半固态的凝胶状。其固含量为15%。所制得的聚酰亚胺前驱物组合物无法涂布成膜,故未量测其涂布性及薄膜性质。
比较例2
在通入氮气的1000毫升的四口反应器中,加入26.431克(0.245摩尔)的对苯二胺、3.554克(0.01摩尔)的9,9’-双(4-氨基苯基)芴及400克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将10.009克(0.046摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将60.005克(0.204摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得比较例2的聚酰亚胺前驱物组合物,但此聚酰亚胺前驱物组合物(即聚酰胺酸)为半固态的凝胶状。其固含量为20%。所制得的聚酰亚胺前驱物组合物无法涂布成膜,故未量测其涂布性及薄膜性质。
比较例3
在通入氮气的1000毫升的四口反应器中,加入19.824克(0.183摩尔)的对苯二胺、2.665克(0.008摩尔)的9,9’-双(4-氨基苯基)芴及425克的n-甲基-2-吡咯烷酮,并于20℃至30℃下混合均匀,以形成二胺化合物溶液。
然后,将7.507克(0.034摩尔)的苯均四羧酸二酐加至此二胺化合物溶液中,以进行初步反应,而可形成经反应的溶液。接着,将45.005克(0.153摩尔)的3,3’,4,4’-二苯基四羧酸二酐均分为三等份,通过分批加入的方式,将其加至前述的溶液中,并持续搅拌,以进行反应。
经过4小时后,即可制得比较例3的聚酰亚胺前驱物组合物(亦即本发明所称的聚酰胺酸)。其粘度约为12000cps,且固含量为15%。所制得的聚酰亚胺前驱物组合物以下述涂布性及薄膜性质的评价方式进行评价,所得结果如表1所示。
制备聚酰亚胺积层板
以下是根据表2制备应用例1至应用例3及比较应用例1的聚酰亚胺积层板。
应用例1
应用例1的聚酰亚胺积层板的制备方法如前所述,在此不另赘述。其中,应用例1的聚酰亚胺前驱物组合物是使用前述实施例1的组合物。所制得的聚酰亚胺积层板以下述薄膜性质与机械性质的评价方式进行评价,所得结果如表2所示。
应用例2及比较应用例1
应用例2及比较应用例1是使用与应用例1的聚酰亚胺积层板相同的制备方法,不同的处在于应用例2是使用前述实施例2的聚酰亚胺前驱物组合物,且比较应用例1是使用比较例3的聚酰亚胺前驱物组合物,且其条件及评价结果如表2所示,在此不另赘述。
评价方式
1.涂布性
聚酰亚胺层的涂布性是将前述实施例1至实施例3及比较例1至比较例2的聚酰亚胺前驱物组合物分别涂布于1/3盎司(厚度约0.012毫米)的铜箔上,并进一步加热进行环化反应,以形成聚酰亚胺层,并依据聚酰亚胺前驱物组合物是否可涂布成膜判断其涂布性。
2.薄膜性质
热性质
聚酰亚胺层的热性质是通过本发明所属技术领域具有通常知识者所熟知的仪器及方法量测前述实施例1至实施例3及比较例1至比较例2所制得的聚酰亚胺层的热膨胀系数(coefficientofthermalexpansion;cte)、玻璃转移温度(tg)及重量损失5%的热裂解温度(td)。其结果如表1所示,在此不另赘述。
尺寸安定性
聚酰亚胺层的尺寸安定性是通过本发明所属技术领域具有通常知识者所熟知的方法量测前述实施例1至实施例3及比较例1至比较例2所制得的聚酰亚胺层于机械方向(machinedirection;md)及横切方向(transversedirection;td)的尺寸安定性。其结果如表2所示,在此不另赘述。
3.机械性质
聚酰亚胺层的机械性质是通过本发明所属技术领域具有通常知识者所熟知的仪器及方法量测前述实施例1至实施例3及比较例1至比较例2所制得的聚酰亚胺层的抗张强度、延伸长度及挠曲性。其结果如表2所示,在此不另赘述。
其中,抗张强度是利用美国电子电路成型标准(associationconnectingelectronicsindustries)中ipc-tm-650第2.4.19号的测试方法量测。挠曲性的破裂测试是以目视的方式观察聚酰亚胺层挠曲后的表面,并依据下述基准进行评价:
○:表面未出现裂纹。
╳:表面出现裂纹。
请参照表1。在聚酰胺酸中,基于二胺化合物的总使用量为100摩尔百分比,当二酐化合物的总使用量不小于98.0摩尔百分比(即比较例1与比较例2)时,所制得的聚酰胺酸易形成半固态的凝胶状,而无法涂布成膜,进而无法制得聚酰亚胺。
请同时参照表1及表2。基于二胺化合物的总使用量为100摩尔百分比,当二酐化合物的总使用量大于或等于95摩尔百分比且小于98摩尔百分比(即实施例1至实施例3时,所制得的聚酰胺酸具有适当的粘度,而可具有良好的涂布性。其中,为了进一步提升所制得的聚酰亚胺的机械性质,实施例1至实施例3导入1摩尔百分比至5摩尔百分比的单体化合物,并通过单体化合物的两个羧基及两个酯基与聚酰胺酸产生反应,而可延长所制得聚酰亚胺的分子链,进而增加其分子量,并提升机械性质。因此,应用例1与应用例2可具有较佳的挠曲性,且具有良好的抗张强度及延伸强度。
于比较应用例1中,基于比较例3的二胺化合物的总使用量为100摩尔百分比,二酐化合物的总使用量为97.9摩尔百分比,但比较例3的聚酰亚胺前驱物组合物并未添加单体化合物。据此,虽然比较例3的聚酰亚胺前驱物组合物(即聚酰胺酸)仍可涂布成膜,但是所制得的聚酰亚胺(即比较应用例1)具有较差的挠曲性。
据此,基于二胺化合物的总使用量为100摩尔百分比时,当二酐化合物的总使用量为本发明前述的范围时,所制得的聚酰胺酸可具有较佳的涂布性。其次,由混合聚酰胺酸与单体化合物可形成本发明的聚酰亚胺前驱物组合物,而可制得具有良好机械性质与薄膜性质的聚酰亚胺。
虽然本发明已以实施方式揭露如上,然其并非用以限定本发明,在本发明所属技术领域中任何具有通常知识者,在不脱离本发明的精神和范围内,当可作各种的更动与润饰,因此本发明的保护范围当视所附的权利要求所界定的范围为准。
- 还没有人留言评论。精彩留言会获得点赞!