一种阿维巴坦钠关键中间体的手性异构体的制备方法与流程
本发明属于医药技术领域,涉及一种阿维巴坦钠关键中间体的手性异构体的方法,具体涉及(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的手性异构体的制备方法。
背景技术:
阿维巴坦钠(Avibactam Sodium,NXL-104)属于二氮杂双环辛酮化合物,由Forest Lab和阿斯利康联合开发,2014年2月阿特维斯收购Forest Lab,取得开发权,2015年2月25日头孢他啶与阿维巴坦钠复方制剂AVYCAZ批准在美国上市。阿维巴坦钠是目前最被看好的新型β-内酰胺酶抑制剂,与三种已上市的β-内酰胺酶抑制剂相比,具有长效和与酶可逆性共价结合,且不会诱导β-内酰胺酶产生。
头孢他啶是一种头孢霉菌素类抗微生物药,对某些革兰氏阴性和革兰氏阳性细菌有体外活性。头孢他啶的杀细菌作用是通过与青霉素结合蛋白(PBPs)结合介导的。阿维巴坦钠是一种非β内酰胺类的β-内酰胺酶抑制剂,灭活有些β-内酰胺酶和保护头孢他啶免受某些β-内酰胺酶降解。
文献所报道的阿维巴坦钠合成主要是以N-叔丁氧羰基-L-焦谷氨酸乙酯为起始物料,经开环、氯代、环合、还原、拆分成盐得关键中间体(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,草酸盐经酰胺化、环合、脱保护、成磺酸四丁基乙酸铵盐、最后经钠盐交换反应得到阿维巴坦钠。(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐为阿维巴坦合成的关键中间体,其结构中有两个手性碳原子,存在三个手性异构体:分别为(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐、(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐、(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐。
在药物开发过程中,中间体的质量控制是一个重要的环节,质量标准的建立需要一定量的对照品,所以杂质的合成方法开发是药物研究的一项重要任务。
技术实现要素:
鉴于此,本发明的目的是通过关键的分离和精制方法,获得阿维巴坦关键中间体(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐中的两个手性异构体(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,为中间体的质量控制提供可靠的杂质对照品。
为了达到上述的目的,本发明的技术方案是这样实现的:
一种阿维巴坦钠关键中间体的手性异构体的制备方法,所述阿维巴坦钠关键中间体为(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,所述手性异构体为(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,所述方法包括以下步骤:
S1、将合成(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液减压浓缩成粘稠物1;
S2、粘稠物1中加入乙酸乙酯和纯化水,调节pH值至碱性,分去水层,有机相干燥、过滤、浓缩得粘稠物2;
S3、粘稠物2经柱层析分离得到(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物;
S4、将混合物溶于丙酮中,滴加草酸二水合物的甲醇溶液,加热搅拌;
S5、降温析晶,过滤除去固体;
S6、滤液加热,再降温析晶,过滤除去固体;
S7、将滤液浓缩后,所得粗品用丙酮和乙醇的混合液中进行重结晶,获得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐精制品。
进一步的,所述步骤S3中,包括以下步骤:
(1)粘稠物2溶于二氯甲烷中,加入200~300目柱层析硅胶拌样,减压蒸干,得拌样样品,其中,粘稠物2与拌样硅胶质量比为1:1~5;
(2)将200~300目柱层析硅胶加入石油醚中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中,其中,粘稠物2和固定相硅胶的质量比为1:10~50;
(3)用石油醚和乙酸乙酯的混合溶剂进行洗脱,收集目标点并减压蒸干,得到游离碱的混合物,其中,石油醚:乙酸乙酯的体积比为1~5:1。
进一步的,所述步骤S4中,加热温度为60~65℃,草酸二水合物和游离碱的摩尔比为0.8~2:1,游离碱:丙酮:甲醇的质量体积比(g/mL/mL)为1:10~30:1。
进一步的,所述步骤S5中的析晶温度为40~50℃;所述步骤S6中,析晶温度为30~40℃。
进一步的,所述步骤S7中的析晶温度为-10~10℃;粗品:丙酮:乙醇的质量体积比(g/mL/mL)为1:1~20:2。
一种阿维巴坦钠关键中间体的手性异构体的制备方法,所述阿维巴坦钠关键中间体为(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,所述手性异构体为(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,所述方法包括以下步骤:
S1、将合成(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液减压浓缩成粘稠物1;
S2、粘稠物1中加入乙酸乙酯和纯化水,调节pH值至碱性,分去水层,有机相干燥、过滤、浓缩得粘稠物2;
S3、粘稠物2经柱层析分离得到(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物;
S4、将混合物溶于乙酸乙酯中,滴加草酸二水合物的甲醇溶液,加热搅拌;
S5、降温析晶,得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯粗品;
S6、粗品于甲苯和甲醇的混合液中加热搅拌,趁热过滤得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯第一次精制品;
S7、第一次精制品于乙酸乙酯和甲醇的混合液中进行重结晶,获得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐精制品。
进一步的,所述步骤S3中,包括以下步骤:
(1)粘稠物2溶于二氯甲烷中,加入200~300目柱层析硅胶拌样,减压蒸干,得拌样样品,其中,粘稠物2与拌样硅胶质量比为1:1~5;
(2)将200~300目柱层析硅胶加入石油醚中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中,其中,粘稠物2和固定相硅胶的质量比为1:10~50;
(3)用石油醚和乙酸乙酯的混合溶剂进行洗脱,收集目标点并减压蒸干,得到游离碱的混合物,其中,石油醚:乙酸乙酯的体积比为1~5:1。
进一步的,所述步骤S4中,加热温度为60~65℃,草酸二水合物和游离碱的摩尔比为0.8~2:1,游离碱:乙酸乙酯:甲醇的质量体积比(g/mL/mL)为1;1~20:1。
进一步的,所述步骤S5中,析晶温度为10~50℃。
进一步的,所述步骤S6中,过滤温度为40~80℃,粗品:甲苯:甲醇质量体积比(g/mL/mL)为1:1~20:2。
进一步的,所述步骤S7中的析晶温度为0~50℃;第一次精制品:乙酸乙酯:甲醇的质量体积比(g/mL/mL)为1:1~20:2。
与现有技术相比,本发明的有益效果为:
本发明通过合成的(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐母液或(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐母液为起始物料,经浓缩、解盐、柱层析、成盐、精制等步骤过程分别得到(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐或(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐。本发明的制备工艺相对简单,纯度高,本发明制备出的手性异构体ee%均为100%。
本发明所制备的手性异构体杂质,对于关键中间体的质量控制,以及成品的质量控制具有重要意义。
附图说明
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单地介绍。
图1为(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的HPLC图谱;
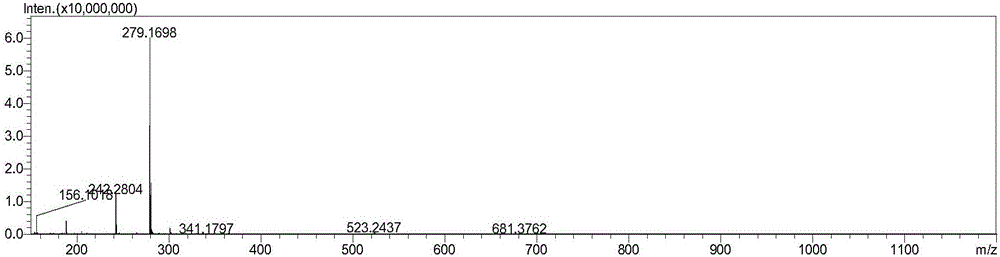
图2为(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的ESI-MS图谱;
图3为(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的1H-NMR图谱;
图4为(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的HPLC图谱;
图5为(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的ESI-MS图谱;
图6为(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的1H-NMR图谱。
具体实施方式
应该指出,以下详细说明都是示例性的,旨在对所要求的本发明提供进一步的说明,除非另有说明,本发明使用的所有技术和科学术语具有与本发明所属技术领域的普通技术人员通常理解的相同含义。
以下结合具体实施例和附图详细说明本发明,目的是使本发明的优点更翔实,而非限制本发明。本领域技术人员应该理解,本发明并不限于这些实施例以及使用的方法,任何对本发明进行同等替换、组合、改良或修饰,均包含于本发明内。
阿维巴坦关键中间体(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的手性异构体之一(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液于40~80℃进行减压浓缩成粘稠物1,优选为40~50℃。
S2、粘稠物1中加入乙酸乙酯和纯化水,用饱和碳酸氢钠水溶液调节pH值至碱性,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得游离碱粘稠物2,其中,pH为7~10,优选为8~9。
S3、粘稠物2经柱层析分离得到(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物2溶于二氯甲烷中,加入200~300目柱层析硅胶拌样,减压蒸干,得拌样样品,其中粘稠物2与拌样硅胶质量比为1:1~5,优选1:1~2;
(2)将200~300目柱层析硅胶加入至石油醚中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中,其中粘稠物2和固定相硅胶的质量比为1:10~50,优选1:10~30;
(3)用石油醚和乙酸乙酯的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中石油醚:乙酸乙酯的体积比为1~5:1,优选2~3:1。
S4、将混合物溶于丙酮中,滴加草酸二水合物的甲醇溶液,加热至60~65℃搅拌30分钟,其中草酸二水合物和游离碱的摩尔比为0.8~2:1,优选0.9~1.1:1,游离碱:丙酮:甲醇的质量体积比(g/mL/mL)为1:10~30:1,优选1:15~25:1。
S5、降温析晶,过滤除去固体,其中析晶温度为40~50℃,优选为40~45℃。
S6、滤液加热,再降温析晶,过滤除去固体,其中析晶温度为30~40℃,优选为30~35℃。
S7、将滤液浓缩后,所得粗品用丙酮和乙醇的混合液进行重结晶,获得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐精制品,其中析晶温度为-10~10℃,优选0~5℃;粗品:丙酮:乙醇的质量体积比(g/mL/mL)为1:1~20:2,优选1:8~12:2。
实施例1
(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液于40~50℃减压浓缩,得16g粘稠物1,HPLC检测(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的质量比为25:75。
S2、粘稠物1中加入90mL乙酸乙酯和50mL纯化水,用饱和碳酸氢钠水溶液调节pH值为8~9,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得11.8g粘稠物2。
S3、粘稠物2经柱层析分离得到(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物2溶解于60mL二氯甲烷中,加入15g 200~300目柱层析硅胶拌样,减压蒸干,得拌样样品;
(2)将250g 200~300目柱层析硅胶加入至1.5L二氯甲烷中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中;
(3)用石油醚:乙酸乙酯的体积比=3:1的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物9g。
S4、将混合物溶于180mL丙酮中,滴加草酸二水合物的甲醇溶液,其中草酸二水合物的添加量为4.4g,甲醇为9mL,加热至60~65℃搅拌30分钟。
S5、降温至40~45℃析晶2小时,过滤除去固体。
S6、滤液加热至50~60℃搅拌30分钟,再降温至30~35℃析晶2小时,过滤,除去固体。
S7、滤液浓缩,得粗品5g,加入丙酮和乙醇的混合液中进行重结晶,获得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,包括以下步骤:
(1)将滤液浓缩后,所得粗品中加入50mL丙酮和10mL乙醇的混合液,加热至50~60℃,维持30分钟;
(2)降温至0~5℃析晶4小时,过滤,所得固体于40~50℃减压干燥得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐3.1g,收率19.4%(以粘稠物1计),ee值100%。
所获得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的HPLC图谱如图1所示;(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的ESI-MS图谱如图2所示;(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的1H-NMR图谱如图3所示。
ESI-MS(m/z):279.16(M+H)
1H-NMR(400MHz,DMSO):δ8.57(br,4H),7.36-7.29(m,5H),4.63(t,2H),4.22-4.12(m,3H),3.16-3.05(m,3H),1.93-1.77(m,2H),1.77-1.74(m,1H),1.48-1.47(m,1H),1.22(t,3H)。
实施例2
(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液于40~50℃减压浓缩,得23.2g粘稠物1。
S2、粘稠物1中加入120mL乙酸乙酯和70mL纯化水,用饱和碳酸氢钠水溶液调节pH值为8~9,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得16.3g粘稠物2。
S3、粘稠物2经柱层析分离得到(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物溶解于100mL二氯甲烷中,加入25g 200~300目柱层析硅胶拌样,减压蒸干,得拌样样品;
(2)将350g 200~300目柱层析硅胶加入至2L二氯甲烷中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中;
(3)用石油醚:乙酸乙酯的体积比=3:1的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物11.3g。
S4、将混合物溶于220mL丙酮中,滴加草酸二水合物的甲醇溶液,其中草酸二水合物的添加量为5.2g,甲醇为11.3mL,加热至60~65℃搅拌30分钟。
S5、降温至40~45℃析晶2小时,过滤除去固体。
S6、滤液加热至50~60℃搅拌30分钟,再降温至30~35℃析晶2小时,过滤,除去固体。
S7、滤液浓缩得粗品6.3g,加入丙酮和乙醇的混合液中进行重结晶,获得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,其中,包括以下步骤:
(1)将滤液浓缩后,所得粗品加入63mL的丙酮和12.6mL乙醇混合液,加热至50~60℃,维持30分钟;
(2)降温至0~5℃析晶4小时,过滤,所得固体于40~50℃减压干燥得(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐4.0g,收率17.2%(以粘稠物1计),ee值100%。
阿维巴坦关键中间体(2S,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的手性异构体之二(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液于40~80℃进行减压浓缩成粘稠物1,优选为40~50℃。
S2、粘稠物中加入乙酸乙酯和纯化水,用饱和碳酸氢钠水溶液调节pH值至碱性,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得粘稠物2,其中pH为7~10,优选为8~9。
S3、粘稠物2经柱层析分离得到(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物2加入200~300目柱层析硅胶拌样,减压蒸干,得拌样样品,其中粘稠物2与拌样硅胶质量比为1:1~5,优选1:1~2;
(2)将200~300目柱层析硅胶加入至石油醚中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中,其中粘稠物2和固定相硅胶的质量比为1:10~50,优选1:10~30;
(3)用石油醚和乙酸乙酯的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中石油醚:乙酸乙酯的体积比为1~5:1,优选2~3:1。
S4、将混合物溶于乙酸乙酯中,滴加草酸二水合物的甲醇溶液,加热至60~65℃搅拌30分钟,其中草酸二水合物和游离碱的摩尔比为0.8~2:1,优选0.9~1.1:1;游离碱:乙酸乙酯:甲醇的质量体积比(g/mL/mL)为1:1~20:1,优选1:8~12:1。
S5、降温析晶,过滤得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐粗品,其中析晶温度为10~50℃,优选为20~30℃。
S6、粗品于甲苯-甲醇中加热搅拌一定时间,趁热过滤得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯第一次精制品,其中过滤温度为40~80℃,优选为50~60℃;其中粗品:甲苯:甲醇质量体积比(g/mL/mL)为1:1~20:2,优选1:10:2;
S7、第一次精制品再于乙酸乙酯和甲醇的混合液中进行重结晶,获得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,其中第一次精制品:乙酸乙酯:甲醇的质量体积比(g/mL/mL)为1:1~20:2,优选1:8~12:2;其中析晶温度为0~50℃,优选20~30℃。
实施例3
(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐后分离得到的母液于40~50℃减压浓缩,得18.1g粘稠物1,HPLC检测(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐质量比为20:80。
S2、粘稠物1中加入150mL乙酸乙酯和100mL纯化水,用饱和碳酸氢钠水溶液调节pH值为8~9,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得12.8g粘稠物2。
S3、粘稠物2经柱层析分离得到(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物溶解于80mL二氯甲烷中,加入20g 200~300目柱层析硅胶拌样,减压蒸干,得拌样样品;
(2)将250g 200~300目柱层析硅胶加入至1.8L二氯甲烷中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中;
(3)用石油醚:乙酸乙酯的体积比=3:1的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物8g。
S4、将混合物溶于80mL乙酸乙酯中,滴加草酸二水合物的甲醇溶液,其中草酸二水合物的添加量为3.6g,甲醇为8mL,加热至60~65℃搅拌30分钟。
S5、降温至20~30℃析晶6小时,得(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐粗品9.5g。
S6、粗品加入至甲苯(95mL)-甲醇(19mL)的混合液中,加热至50~60℃搅拌2小时,趁热过滤、干燥得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯第一次精制品6.2g。
S7、将第一次精制品加入至乙酸乙酯和甲醇的混合液中进行重结晶,获得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,其中,包括以下步骤:
(1)第一次精制品加入至62mL的乙酸乙酯和12.4mL甲醇混合液中,加热至50~60℃,维持30分钟;
(2)降温至20~30℃析晶4小时,过滤,所得固体于40~50℃减压干燥得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐4.7g,收率26.0%(以粘稠物1计),ee值100%。
所获得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的HPLC图谱如图4所示;(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的ESI-MS图谱如图5所示;(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的1H-NMR图谱如图6所示。
ESI-MS(m/z):279.16(M+1)
1H-NMR(400MHz,DMSO):δ7.36-7.29(m,5H),6.98(br,4H),4.62(t,2H),4.20-4.14(m,2H),3.98-3.97(m,1H),3.07(d,2H),3.02-2.98(m,1H),1.89-1.82(m,2H),1.73-1.72(m,1H),1.48-1.47(m,1H),1.21(t,3H)。
实施例4
(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐的制备,包括以下步骤:
S1、将合成的(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐母液于40~50℃减压浓缩,得10.2g粘稠物1。
S2、粘稠物1中加入80mL乙酸乙酯和50mL纯化水,用饱和碳酸氢钠水溶液调节pH值为8~9,分去水层,有机层用无水硫酸钠干燥、过滤、浓缩得7.2g粘稠物2。
S3、粘稠物2经柱层析分离得到(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物,其中,包括以下步骤:
(1)粘稠物溶解于50mL二氯甲烷中,加入10g 200~300目柱层析硅胶拌样,减压蒸干,得拌样样品;
(2)将160g 200~300目柱层析硅胶加入至1.8L二氯甲烷中,搅拌均匀后加入至玻璃层析柱中,沉降,加压将柱子压实,将拌样样品加入至玻璃层析柱中;
(3)用石油醚:乙酸乙酯的体积比=3:1的混合溶剂进行洗脱,收集目标点并减压蒸干,得(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱和(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯游离碱的混合物5.2g。
S4、将混合物溶于52mL乙酸乙酯中,滴加草酸二水合物的乙醇溶液,其中草酸二水合物的添加量为2.4g,乙醇为5.2mL,加热至60~65℃搅拌30分钟。
S5、降温至20~30℃析晶6小时,过滤得(2R,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐固体6.2g。
S6、粗品加入至甲苯(62mL)-甲醇(12.4mL)的混合液中,加热至50~60℃搅拌2小时,趁热过滤、干燥得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯第一次精制品3.8g。
S7、将第一次精制品加入至乙酸乙酯和甲醇的混合液中进行重结晶,获得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐,其中,包括以下步骤:
(1)第一次精制品加入至38mL乙酸乙酯和7.6mL甲醇混合液中,加热至50~60℃,维持30分钟;
(2)降温至20~30℃析晶4小时,过滤,所得固体于40~50℃减压干燥得(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐2.8g,收率27.5%(以粘稠物1计),ee值100%。
本发明的制备工艺相对简单,通过本发明所述的方法制备的阿维巴坦钠关键中间体的手性异构体(2S,5S)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐或(2R,5R)-5-(苄氧氨基)-哌啶-2-甲酸乙酯草酸盐纯度高ee值为100%。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!