一种抗体药物偶联物中间体及其制备方法与流程
本发明涉及药物化学领域,特别涉及一种可以与单克隆抗体偶联的抗体药物偶联物中间体及其制备方法。
背景技术:
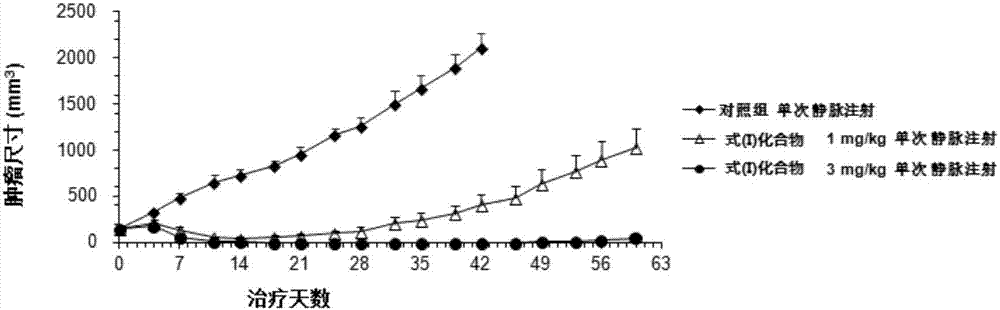
:抗体药物偶联物(adc)是一类新的,很有前途的靶向性治疗各种恶性肿瘤的化疗药物。adc包括三个不同的部分,即抗体、连接链和细胞毒素,实现了将细胞毒素向癌细胞的有效靶向递送。其机制是通过adc中的抗体与肿瘤特异性过表达的细胞表面抗原结合后,经过内吞来实现运送细胞毒素到特定的肿瘤细胞内,从而提高了细胞毒素的功效和选择性。与传统的化疗药物相比,adc以癌细胞作为靶向细胞并特定性地攻击癌细胞,对健康细胞较少造成严重的影响。由于可以运送到肿瘤细胞内的细胞毒素分子的数量有限,只有高度有效的细胞毒素才可被用作有效毒素使用。duostatin5是一种基于有高度细胞毒性的天然产物dolastatin10而设计的细胞毒素,它能满足在adc中作为有效细胞毒素的需要。目前在adc类药物的制备过程中,连接链采用缬氨酸-胍氨酸形成的可被肽酶裂解的二肽,并将fmoc-缬氨酸-胍氨酸作为关键组分和毒素在酰胺化条件下反应。但是由于胍氨酸非常容易成环,所以反应收率一般。同时因为胍氨酸在肽偶合条件下容易发生异构化,产物的光学纯度低。合成过程中产生的不需要的非对映异构体需要通过繁琐的反相hplc纯化分离。技术实现要素:针对现有技术存在的缺陷,本发明提供一种操作简单,容易纯化,能减少和规避酰胺化步骤中胍氨酸差向异构化,以及胍氨酸衍生物在有机溶剂中的低溶解性问题的工艺路线及采用本发明的路线制备的抗体药物偶联物中间体。因此,本发明的一个目的是提供一种抗体药物偶联物中间体。本发明的另一目的是提供一种制备上述抗体药物偶联物中间体的方法。实现本发明目的的技术方案如下:一方面,本发明提供一种抗体药物偶联物中间体,该中间体为式(i)所示的化合物、对映异构体、外消旋体或其药学上可接受的盐:另一方面,本发明提供一种制备上述药物偶联物中间体的方法。该方法操作简单,产品容易纯化,并能减少和规避酰胺化步骤中胍氨酸差向异构化,同时解决了胍氨酸衍生物在有机溶剂中的低溶解性问题。为了达到这个目的,我们设计了以下技术方案:具体地,上述抗体药物偶联物中间体的制备方法包括以下步骤:1)将化合物1或其盐与化合物2溶解于溶剂中,在试剂a的作用下经缩合反应得到化合物3;2)将化合物3从步骤1)得到的反应液中分离;3)将步骤2)得到的化合物3溶解于溶剂中,在试剂b的作用下脱掉氨基保护基l,得到化合物4;4)将化合物4从步骤3)得到的反应液中分离;5)步骤4)得到的化合物4与化合物5在试剂c的作用下经缩合反应得到化合物6;6)将化合物6从步骤5)得到的反应液中分离;7)步骤6得到的化合物6溶解于溶剂中,在试剂d的作用下,脱去氨基保护基m,得到化合物7;8)将化合物7从步骤7)得到的反应液中分离;9)将步骤8)得到的化合物7溶解于溶剂中,调节溶液ph,在试剂e的作用下,得到化合物8;10)将步骤9)得到的化合物8与试剂f反应,得到化合物9;11)将化合物9从步骤10)得到的反应液中分离;12)将步骤11)得到化合物9溶解于溶剂中,并与试剂g反应,得到式(i)所示的化合物;13)将式(i)所示的化合物从步骤12)得到的反应液中分离;其中化合物2、3、4、6中的m和l为氨基保护基,且m与l不同。优选地,在步骤1)中,所述溶剂选自二氯甲烷、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四氢呋喃、1,4-二氧六环和2-甲基四氢呋喃中的一种或多种;更优选地,所述溶剂选自二氯甲烷和n,n-二甲基甲酰胺中的一种或多种。优选地,在步骤1)中,所述试剂a选自dcc、edc、dic、hatu、hbtu、hbpipu、hbpyu、hspyu、hctu、hotu、hott、hstu、hdma、tatu、tbtu、tctu、tcfh、tdbtu、totu、tott、tptu、tffh、btffh、tntu、tstu、comu、t3p、bop、pybop、pybrop、pyclop、brop、pyaop、pyciu、cdi、对甲基苯磺酰咪唑、tpsi、tstu、depbt、dmtmm、eedq、cip、cib、hoat、hobt和dmc中的一种或多种;更优选地,所述试剂a选自edc、dic、hoat和hobt中的一种或多种;进一步优选地,所述试剂a为edc或dic与hoat或hobt的混合物;优选地,在步骤1)中,所述试剂a作为最后一个组分加入到反应液中。优选地,在步骤1)中,反应温度为-20℃-100℃,优选为15℃-30℃。优选地,在步骤1)中,所述氨基保护基m为cbz、boc(叔丁基氧羰基)、troc或tr,优选为boc。优选地,在步骤1)中,所述氨基保护基l为fmoc或sem,优选为fmoc。优选地,在步骤2)中,所述分离包括将所述反应液用等体积的水或饱和食盐水洗涤,并蒸去溶剂,以得到化合物3。优选地,在步骤3)中,所述溶剂选自dmf、dmso、二氯甲烷和乙腈中的一种或多种;更优选地,所述溶剂为乙腈。优选地,在步骤3)中,所述试剂b选自但不限于dbu、哌啶、乙二胺、二异丙胺、吗啉、n-甲基哌嗪和吡咯烷中的一种或多种;更优选地,所述试剂b选自哌啶和二异丙胺中的一种或多种,最优选为哌啶。优选地,在步骤3)中,反应温度为0℃-100℃,更优选为15℃-30℃。优选地,在步骤4)中,所述分离包括将步骤3)得到的反应液蒸发和干燥,剩余的粗品再溶解在二氯甲烷;化合物4的二氯甲烷溶液用0.1-0.5mhcl水溶液洗涤到为水相且最终水相的ph为2-5;水溶液再用二氯甲烷洗涤;然后水溶液用碳酸氢钠或其它无机碱中和到ph为8-10。碱化后的水溶液用二氯甲烷萃取,得到化合物4的二氯甲烷的溶液;二氯甲烷萃取液再用水或饱和食盐水洗涤,干燥后蒸发得到化合物4。优选地,在步骤5)中,所述试剂c选自dcc、edc、dic、hatu、hbtu、hbpipu、hbpyu、hspyu、hctu、hotu、hott、hstu、hdma、tatu、tbtu、tctu、tcfh、tdbtu、totu、tott、tptu、tffh、btffh、tntu、tstu、comu、t3p、bop、pybop、pybrop、pyclop、brop、pyaop、pyciu、cdi、对甲基苯磺酰咪唑、tpsi、tstu、depbt、dmtmm、eedq、cip、cib、hoat、hobt、hatu、dipea和dmc中的一种或多种;更优选地,所述试剂c选自edc、dic、hoat和hobt中的一种或多种;进一步优选地,所述试剂c为edc或dic与hoat或hobt的混合物;优选地,在步骤5)中,所述试剂c作为最后一个组分加入到反应液中。优选地,在步骤5)中,反应温度为-20℃-100℃,优选为15℃-30℃。优选地,在步骤6)中,所述反应液用等体积的水或饱和食盐水洗涤,并蒸去溶剂,得到化合物6。优选地,在步骤7)中,所述溶剂选自1,4-二氧六环、乙酸乙酯、甲醇、乙醇和异丙醇中的一种或多种;更优选地,所述溶剂为1,4-二氧六环。优选地,在步骤7)中,所述试剂d选自h2so4、hcl和tfa中的一种或多种;更优选地,所述试剂d为hcl。优选地,在步骤8)中,所述分离包括所述反应液在减压下除去溶剂并干燥得到化合物7。优选地,在步骤9)中,所述溶剂选自甲醇、乙醇、异丙醇和水中的一种或多种;更优选地,所述溶剂为乙醇和水的混合溶剂。优选地,在步骤9)中,所述试剂e为氰酸盐,如氰酸钠、氰酸钾、氰酸三取代胺盐、氰酸四取代胺盐等;更优选地,所述试剂e为氰酸钠。优选地,在步骤9)中,所述ph值为4-9;更优选地,所述ph值为5-7。优选地,在步骤9)中,所述ph采用酸碱形成的缓冲溶液来调节,所述缓冲溶液可以是有机碱与酸形成的缓冲溶液,如三甲基胺、三乙基胺、三丁基胺、二异丙基乙基胺与盐酸、醋酸、三氟乙酸形成的缓冲溶液;或者是碱与其相应的酸形成的缓冲溶液,如醋酸钠、醋酸钾与醋酸,磷酸二氢钾、磷酸二氢钠与磷酸氢二钾、磷酸氢二钠或者磷酸二氢钾、磷酸二氢钠与磷酸形成的缓冲溶液。更优选地,所述ph采用三乙基胺和醋酸形成的缓冲溶液调节。优选地,在步骤10)中,所述试剂f为无机碱,如氢氧化钠、氢氧化钾、氢氧化锂、氢氧化铯、碳酸钾、碳酸钠、碳酸锂和碳酸铯等。更优选地,所述试剂f为氢氧化钠或氢氧化锂。优选地,在步骤10)中,反应温度为10℃-80℃,优选为40℃-55℃。优选地,在步骤11)中,所述分离采用反相-hplc;更优选地,选用gemininx5u,c18,柱,乙腈(ch3cn)/水体系作为洗脱液,三氟乙酸作为添加剂。优选地,在步骤12)中,所述化合物9与所述试剂g是在缩合剂,如dcc、dic或edc,优选edc,存在下发生的反应。优选地,在步骤12)中,所述溶剂选自二氯甲烷、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、四氢呋喃、1,4-二氧六环和2-甲基四氢呋喃中的一种或多种;更优选地,所述溶剂为二氯甲烷和n,n-二甲基甲酰胺中的一种或多种。优选地,在步骤12)中,所述试剂g选自但不限于n-羟基琥珀酰亚胺、五氟苯酚、硝基酚或其类似物或衍生物;更优选地,所述试剂g为五氟苯酚。优选地,在步骤12)中,反应温度为-20℃-100℃,优选为15℃-30℃。优选地,在步骤13)中,所述分离采用反相-hplc;更优选地,选用gemininx5u,c18,柱,乙腈(ch3cn)/水体系作为洗脱液,三氟乙酸作为添加剂。如本文所用,常用的缩写及其定义如表1所示:表1缩写定义缩写定义aq.水溶液tlc薄层层析boc或boc叔丁基氧羰基μl微升troc三氯乙基氧羰基ms质谱tr三苯甲基pab对氨基苄基fmoc9-芴甲基氧羰基rp-hplc反相高效液相sem2-三甲基硅基乙氧甲基氧羰基lc/ms液质联用hplc高效液相eq当量ml毫升℃摄氏度g克h小时如本文所用,常用的有机物缩写的定义及其相应的cas号如表2所示:表2与抗体偶联的本发明的抗体药物偶联物中间体在针对多种肿瘤细胞株的体外细胞杀伤实验中被证明是非常有效的,在各种动物模型中能有效地抑制肿瘤生长。本发明还涉及一种抗体药物偶联物中间体及其改进的、更有效的适合于大规模生产的制备方法。本发明的方法提高了抗体药物偶联物中间体的收率,且易于提纯。本发明的关键是在合成中利用鸟氨酸作为合成胍氨酸的前体来减少和规避酰胺化步骤中胍氨酸差向异构化的问题,以及胍氨酸衍生物在有机溶剂中的低溶解性问题。在合成的非常晚期的阶段以定量的方式将鸟氨酸转化为胍氨酸。附图说明图1:在jimt-1体内研究中,肿瘤体积随治疗天数的变化;图2:在jimt-1体内研究中,小鼠体重随治疗天数的变化;图3:在hcc1954体内研究中,肿瘤体积随治疗天数的变化;图4:在hcc1954体内研究中,小鼠体重随治疗天数的变化;图5:化合物9的核磁图谱。具体实施方式下面结合具体实施方式对本发明的技术方案作进一步非限制性的详细说明。需要指出的是,下述实施例仅为说明本发明的技术构思及特点,其目的在于让熟悉此项技术的人士能够了解本发明的内容并据以实施,并不能以此限制本发明的保护范围。凡根据本发明精神实质所作的等效变化或修饰,都应涵盖在本发明的保护范围之内。实施例1:化合物1的制备1.1tra的制备将化合物ii(1.3kg,3.55mol)溶于dcm(3l),原料不能完全溶解,降温至0℃,加入et3n(718g,7.10mol)原料全部溶解,滴加mscl(447g,3.89mol),保持温度在5℃以下,反应2h,点板检测反应完全,反应液用水,食盐水洗涤后干燥,过滤,浓缩,得化合物iii1.6kg(收率101%)。将化合物iii(1.6kg,3.6mol),溶于dmf(12l),加入nan3(1.2kg,18mol)加热至65℃,反应3h,点板检测原料大部分反应掉,且有副产物产生,终止反应,将反应液加入到冰水中,用乙酸乙酯萃取,有机相用食盐水洗涤,干燥,过滤,浓缩的粗品,过柱纯化(pe:ea=10:1),得到化合物iv400g(收率28.4%)。将化合物iv(400g,1.02mol)溶于1l1,4-二氧六环中降温至0℃,加入3l6mhcl-1,4-二氧六环,室温反应过夜,lcms检测反应完全,将反应液加到5l甲基叔丁基醚中,产品析出,过滤,将滤饼干燥,得化合物v204g(收率75.8%)。将化合物v(204g,0.772mol)溶于水(2l),降温至0℃,加入na2co3调节ph到中性,加入acoh调节ph=3-4,加入fmoc-cl(200g,0.773mol)的thf(1l)溶液,反应室温过夜,lcms显示反应完全,加入na2co3调节ph=8-9,乙酸乙酯(ea)萃取,有机相合并后用水洗涤。有机相干燥过滤浓缩得粗品,过柱纯化(dcm:meoh=150:1),得tra180g(收率56.4%)。1.2化合物1的制备化合物1采用如下路线制备:具体步骤如下:将化合物1-1(48g,167.04mmol,合成方法参考tetrahedronp913-1924.1993),tra(76.74g,185.60mmol)溶于1.5l的dcm中,冰浴降温至0℃,加入edci(96g,501.13mmol),hobt(22.57g,167.04mmol),在n2保护下室温反应过夜,反应液用水洗涤一次,柠檬酸洗涤两次,na2co3水溶液洗涤两次,食盐水洗涤,有机相干燥过滤浓缩得粗品110g,粗品用2l的ea溶解,过滤掉不溶物,加入8l正己烷,将混合溶剂加热至澄清,静置降温结晶,过滤得滤饼(即化合物1-2)80g(收率70%)。将化合物1-2(80g)溶于400mldcm中,降温至0℃,加入6mhcl-1,4-二氧六环150ml,室温反应4h,lcms检测原料反应完全,浓缩,溶剂拉干得产品(即化合物1-3)70g.直接用于下一步反应。将化合物1-3(135g,218mol),dmt(132.7g,284.71mol,合成方法参考chemicalandpharmaceuticalbulletin,1995,v43,p1706-1718)溶于1.5l的dcm中,降温至0℃,n2保护。加入dipea(85g,657.03mol),hobt(59.2g,438.02mol),edci(84g,438.02mol),0℃下搅拌30分钟,升温至25℃反应过夜。lcms检测原料反应完全。溶剂旋干。将粗品溶于柠檬酸水溶液中(4l)。水相用mtbe(2l*3)萃取。点板至产品点上方杂质点消失。调节水相ph=8-9,ea萃取。有机相干燥过滤浓缩得产品210g,hplc检测纯度94%。重结晶得到产品(即化合物1-4)175g(收率80.8%)。将化合物1-4(180g,181.04mol)溶于1l的dcm,dmf(300ml)中,降温至0℃,加入dipa(300ml),25℃下反应12-15小时,lcms检测反应完全。溶剂拉干,粗品用500ml的dcm溶解,滴加到n-hexane:dcm=16l:400ml中,重复两次,过滤,滤饼烘干得125g产品(即化合物1)(收率89.4%)。采用核磁共振氢谱对所得产品进行结构确证,结果如下:1hnmr(400mhz,chloroform-d)ppm0.81-0.88(m,3h)0.91-1.12(m,18h)1.16-1.45(m,6h)1.67-1.95(m,7h)1.98-2.14(m,4h)2.24(br.s.,1h)2.27-2.56(m,10h)3.05(s,2h)3.32-3.60(m,12h)3.90(d,j=8.07hz,1h)4.01-4.18(m,2h)4.18-4.32(m,1h)4.81(dd,j=8.80,6.85hz,1h)6.63(d,j=8.31hz,2h)6.91-7.05(m,3h)。实施例2:本实施例的反应路线如下:2.1化合物3的制备依次将化合物1(5克,6.48mmol,1.0eq.)、化合物2(吉尔生化,2.95克,1.0eq.)、hobt(共价化学,2.3g,3.0eq.)、二氯甲烷(200毫升)和二异丙基乙基胺(共价化学,5.9毫升,6eq.)加入到反应瓶中。搅拌溶解成澄清溶液。然后加入edc(3.23克,3eq.),在室温下搅拌1-2小时。lcms检测反应完毕。反应液用水(2×50毫升)、饱和食盐水(50毫升)洗涤。除去溶剂后得到7.64克化合物3。并采用核磁共振氢谱对其结构进行确证,结果如下:1hnmr(400mhz,chloroform-d)ppm0.83-1.12(m,19h)1.17-1.25(m,3h)1.26-1.37(m,10h)1.38-1.49(m,10h)1.52(br.s.,1h)1.72(br.s.,12h)1.82-1.95(m,3h)1.95-2.16(m,4h)2.25(d,j=7.58hz,2h)2.29-2.52(m,6h)2.87(br.s.,2h)3.07(br.s.,3h)3.16(br.s.,1h)3.24-3.46(m,8h)3.46-3.58(m,3h)3.86(d,j=4.16hz,1h)4.17-4.37(m,3h)4.42(br.s.,2h)4.68-4.86(m,2h)7.11-7.23(m,2h)7.29-7.37(m,2h)7.41(t,j=7.21hz,2h)7.52(br.s.,2h)7.62(br.s.,2h)7.78(d,j=7.34hz,2h)。2.2化合物4的制备将化合物3(7.6g)溶解在70毫升的乙腈溶液中,加入4毫升的哌啶,在室温下搅拌20分钟。lcms检测反应完成。反应完成后,减压下将溶剂蒸发;残留物溶解在50毫升乙腈并再次蒸发,残留物再次用50毫升乙腈溶解并再次蒸发,然后溶于二氯甲烷,用水洗2次。二氯甲烷溶液用0.5m盐酸萃取(hcl水溶液,约需要2.5-3产品当量的盐酸,萃取液ph值必须为4或更低)。萃取的水溶液用二氯甲烷反萃取一次。萃取的水溶液用碳酸氢钠碱化至ph值为9。用二氯甲烷萃取碱化后的混合物水溶液,二氯甲烷萃取液用水洗两次,然后蒸干得到4.8克化合物4。并采用核磁共振氢谱对其结构进行确证,结果如下:1hnmr(400mhz,chloroform-d)dppm0.75-1.03(m,17h)1.04(br.s.,3h)1.11(d,j=6.60hz,1h)1.20(d,j=6.60hz,3h)1.24-1.36(m,4h)1.57-1.83(m,7h)1.86-2.16(m,7h)2.23-2.38(m,8h)2.44-2.64(m,2h)2.79-2.91(m,2h)3.05(s,2h)3.12-3.29(m,4h)3.29-3.53(m,10h)3.53-3.64(m,1h)3.87(br.s.,1h)4.07(br.s.,1h)4.80(t,j=7.58hz,1h)6.97(br.s.,1h)7.11(d,j=8.31hz,1h)7.19(m,j=8.07hz,2h)7.47-7.64(m,2h)。2.3化合物6的制备依次将化合物4、化合物5(豪晶化学,1.0eq.)、hobt(共价化学,3.0eq.)、二氯甲烷(200ml)和二异丙基乙基胺(4eq.)加入到反应瓶中。搅拌溶解成澄清溶液。然后加入edc(共价化学,3eq.),在室温下搅拌1-2小时。lcms检测反应完毕。反应液用水(2×50ml)、饱和食盐水(50ml)洗涤。除去溶剂后得到6.4g化合物6。并采用核磁共振氢谱对其结构进行确证,结果如下:1hnmr(400mhz,chloroform-d)dppm0.83-1.12(m,27h)1.14-1.33(m,15h)1.46(br.s.,13h)1.67(br.s.,21h)1.75(br.s.,3h)2.24(s,1h)2.48(d,j=12.72hz,2h)3.06(s,2h)3.19-3.42(m,8h)3.42-3.59(m,3h)3.59-3.80(m,4h)4.80(br.s.,1h)7.02-7.20(m,2h)7.77(d,j=7.34hz,2h)。2.4化合物7的制备将上一步得到的化合物6溶解在15毫升甲醇中,在搅拌下加入50毫升4mhcl的1,4-二氧六环溶液。20分钟后lcms检测反应完成。反应完成后,减压下蒸发溶剂,残留物溶解在50ml乙腈并再次蒸发,残留物再次用50ml乙腈溶解并再次蒸发,残留物在真空下干燥至少1h,得到化合物7。2.5化合物8的制备将上一步得到的化合物7溶解在25毫升乙醇和25毫升水中。用二异丙基乙基胺调节ph到5-6(如果加了过量的二异丙基乙基胺,可以再加入醋酸ph值),然后加入氰酸钠(3eq.),搅拌16小时。有一些沉淀物形成。lcms检查反应完成。如果反应没有完成,检查ph值(ph值应在5和7之间)。反应完成后,进入下一步。2.6化合物9的制备向上一步得到的中间体8的反应混合液中加入10eq.的naoh(国药,溶解在3毫升的水中)。在搅拌下将反应混合物温度升至50℃。1小时后lcms检测反应完成。反应完成后用醋酸调节ph值至5,然后用高效液相色谱法纯化并冻干相应的组分得到化合物9(4.17克,3.36mmol):制备hplc条件:柱子:gemininx5u,c18,110a,150x50mm流动相:乙腈/0.1%三氟乙酸水溶液梯度:20分钟,从5%到55%乙腈流速:50毫升/分钟产品在17-19分钟时洗脱并采用核磁共振氢谱对得到的化合物9进行结构进行确证,结果如图5所示。2.7式(i)化合物的制备将化合物9的tfa盐(4.17克,3.12mmol)溶解在100ml二氯甲烷(如果不能完全溶解,加入总共不超过10毫升n,n-二甲基甲酰胺助溶)。加入五氟苯酚(sigma-aldrich,5eq.),然后添加edc(共价化学,3eq.)。搅拌20分钟后lcms检查反应是否完成。如果没有完成,添加更多的edc,搅拌10-20分钟后再次检查。反应完成后,用80毫升的水洗涤,二氯甲烷溶液用na2so4干燥约2-5分钟,然后在室温下减压下蒸发溶剂(注:洗涤步骤应该做的快,几分钟内完成以避免水解)。残留物用乙腈/水溶解(7/3体积比),用高效液相色谱分10-15次纯化。每次注射约0.5-0.7克的混合物:制备hplc条件:柱子:gemininx5u,c18,110a,150x50mm流动相:乙腈/0.1%三氟乙酸水溶液梯度:20分钟,从10到65%乙腈流速:50毫升/分钟产品在18-20分钟时洗脱在主峰左边,右边的边峰,以及主峰的组分用分析高效液相色谱法分析(柱子:gemini--nx-5u,c18,4.6x150mm,ch3cn-h2o(0.1%tfa)20-70%,16min,uv220nm,~出峰时间大约在9.5分钟)。(注:为了避免在高效液相色谱分析过程中,对水分敏感的活性酯被水解。首先采集样品用于hplc分析,然后将各组分分别冷冻和冻干。冷冻干燥后,所需的合适组分可以合并)将含有所需的非对映异构体(主要的非对映异构体)和可接受纯度的组分合并后得到3.8克白色粉末状的式(i)化合物。产率:从化合物1计算44%。lcms(柱子:phenomenexgemininx,5u,c18,50x4.6mm,乙腈/水(0.1%甲酸)5~95%,5min)msm/z1405.5(m+h)。并采用核磁共振氢谱对其结构进行确证,结果如下:1hnmr(400mhz,dmso-d6)dppm0.78(q,j=6.93hz,3h)0.83-1.09(m,26h)1.24(br.s.,4h)1.27-1.41(m,3h)1.47(br.s.,3h)1.65(d,j=11.98hz,1h)1.73(br.s.,1h)1.79-1.95(m,3h)1.96-2.13(m,2h)2.22-2.36(m,2h)2.39(br.s.,1h)2.54-2.71(m,5h)2.77(td,j=7.76,4.28hz,8h)2.98-3.08(m,4h)3.08-3.25(m,7h)3.25-3.44(m,4h)3.48(br.s.,1h)3.61-3.86(m,2h)3.86-4.15(m,5h)4.31(br.s.,1h)4.53-4.76(m,2h)6.01(br.s.,1h)6.98-7.17(m,2h)7.53-7.74(m,2h)7.87(td,j=16.38,8.31hz,1h)8.02-8.31(m,1h)8.93-9.11(m,1h)9.54(br.s.,1h)实施例3:化合物3的制备将化合物1(500mg,0.65mmol,1.0eq.),化合物2(323.80mg,0.712mmol,1.1eq.)在n2保护下,反应1h,lcms检测反应,反应完全。有机相用na2co3水溶液洗涤,干燥,过滤,浓缩得粗品,粗品过柱纯化(dcm:meoh=50:1)得600mg的化合物3。实施例4:化合物4的制备将化合物3(600mg)溶于乙腈,加入1ml哌啶,反应1h,lcms检测反应完全。浓缩,固体用少量dcm溶解,滴加到正己烷中,反复3次,过滤得产品(即化合物4)200mg。实施例5:化合物4的制备将化合物3(2g,1.65mmol)溶于20ml二氯甲烷中,加入2mldmf,降温至0℃,加入5ml二异丙胺,30℃反应4h,lc-ms检测反应完全,将溶剂拉干,用10mldcm溶解,滴加到600ml正己烷中,重复两次,过滤,滤饼烘干得产品(即化合物4)1.3g。实施例6:化合物6的制备将化合物4(1g,1.01mmol,1eq),化合物5(416mg,1.22mmol,1.2eq),n2保护,降温至0℃,加入hatu(578mg,1.52mmol,1.5eq),dipea(262mg,2.03mmol,2eq),室温反应2h,lcms检测反应完全,浓缩,粗品用乙酸乙酯溶解,有机相用na2co3溶液洗涤,食盐水洗涤,干燥,过滤。浓缩得粗品,(hplc检测有10%消旋体),过柱纯化得300mg纯产品(即化合物6)。实施例7:化合物8的制备1.配制ph=5.5hoac-naoac缓冲溶液待用,取0.9克化合物7的粗品,溶于4ml乙醇,加入2ml水,磁力搅拌,再加入配制好的2mlhoac-naoac缓冲溶液,溶液ph值在5~6之间,再加入0.074克氰酸钠,20℃反应过夜14小时后,取样检测(lc-ms,hplc),原料剩余12%左右,补加2mlhoac-naoac缓冲溶液,0.03克氰酸钠,继续反应48小时后,原料无剩余,结束反应。2.反应液未进一步纯化,直接用于下一步反应。实施例8:对jimt-1肿瘤生长的抑制jimt-1肿瘤细胞在体外单层培养。当细胞处于一个指数生长期时收获,计数肿瘤并接种。在每只小鼠在右下侧面皮下接种0.2毫升的无血清培养基jimt-1肿瘤细胞(10×106)并让肿瘤发展。接种肿瘤7天后,当肿瘤平均体积达164立方毫米时开始治疗。将带肿瘤小鼠按照肿瘤体积和体重,分为不同的组(n=8)。按图例所示给药,对照组注射的是t-dm1(购买自罗氏制药),经静脉注射对小鼠进行给药。每周两次用游标卡尺按两个维度测量肿瘤的大小,肿瘤的大小用公式v=0.5a×b2计算,用立方毫米(mm3)表示,其中分别是a和b是肿瘤的长径和短径。其中图1表示,在jimt-1体内研究中,肿瘤体积随治疗天数的变化;图2表示,在jimt-1体内研究中,小鼠体重随治疗天数的变化。实施例9:对hcc1954肿瘤生长的抑制hcc1954肿瘤细胞在体外单层培养。当细胞处于一个指数生长期时收获,计数肿瘤并接种。每只小鼠在右下侧面皮下接种0.2毫升的无血清培养基和基质胶(1:1)hcc1954肿瘤细胞(5×106)让肿瘤发展。接种肿瘤12天后,当肿瘤平均体积达168立方毫米时开始治疗。将带肿瘤小鼠按照肿瘤体积和体重,分为不同的组(n=8)。按图例所示给药,对照组注射的是t-dm1(购买自罗氏制药),经静脉注射对小鼠进行给药。每周两次用游标卡尺按两个维度测量肿瘤的大小,肿瘤的大小用公式v=0.5a×b2计算,用立方毫米(mm3)表示,其中分别是a和b是肿瘤的长径和短径。其中图3表示,在hcc1954体内研究中,肿瘤体积随治疗天数的变化;图4表示,在hcc1954体内研究中,小鼠体重随治疗天数的变化。实施例10:化合物9的制备采用如下路线制备化合物9:化合物12的制备:将化合物10(261mg,0.52mmol)溶解于6mldmf,然后将hatu(217mg,0.57mmol),diea(362l,2.08mmol),和化合物11(213mg,0.52mmol)加入到化合物10的溶液中。所得的混合物在室温下搅拌30分钟。然后向反应液中加入400l哌啶,继续搅拌10分钟。除去溶剂,残留物用hplc纯化得到化合物12(171mg,60%)。msm/z548.3(m+h)。化合物14的合成:将化合物12(37mg,0.15mmol)溶解于4mldmf。然后将hatu(59mg,0.15mmol),diea(108l,0.6mmol),和化合物13(102mg,0.15mmol)加入到化合物12的溶液。所得的反应液在室温下搅拌30分钟,然后蒸干。残余物溶解于2mldcm,然后加入1mltfa并搅拌10分钟。混合物蒸干并用hplc纯化得到化合物14。(94mg,78%)。msm/z673.4(m+h)。化合物9的合成:将化合物14(85mg,0.12mmol)溶解于2mldmf,然后将hatu(48mg,0.12mmol),diea(83l,0.48mmol),和化合物15(94mg,0.12mmol)加入到化合物13的溶液中。所得的反应液在室温下搅拌30分钟,然后加入90mgnaoh溶解于1ml水的溶液并继续搅拌30分钟反应液用hplc纯化得到化合物9(86mg,58%)。msm/z1239.7(m+h)。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1