一种猪嵴病毒全长cDNA的构建方法与流程
本发明涉及生物
技术领域:
,具体涉及一种猪嵴病毒全长cDNA的构建方法。
背景技术:
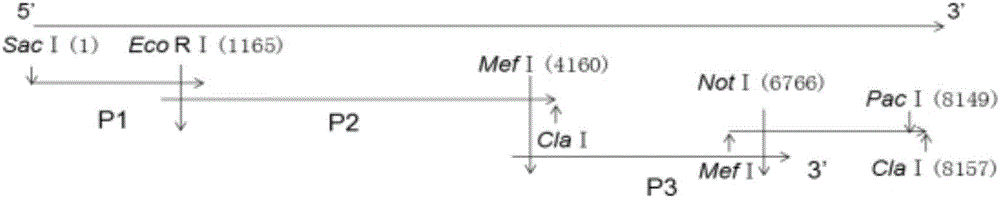
:近年来,反向遗传操作技术在病毒学研究中取得了一定进展。构建感染性cDNA后通过体内或体外的方式获得病毒RNA的方法,在RNA病毒的研究中将发挥着越来越重要的作用。对病毒基因进行修饰拯救出具有生物活性的病毒后将其与野生型病毒进行比较,能为病毒病的防制及疫苗研究提供思路。猪嵴病毒的研究进程因未找到合适的病毒分离及纯化方法而受到严重阻碍。技术实现要素:鉴于现有技术的缺陷,本发明旨在于提供一种猪嵴病毒全长cDNA的构建方法。为获的纯化的病毒,本发明通过分析从流行病学调查中选出的在腹泻仔猪中较流行的PKVCH441株基因组和pBlueScriptKS(Ⅱ)载体结构,对PKVCH441株基因组进行了分段RT-PCR扩增并克隆在pBlueScriptKS(Ⅱ)载体上,成功构建了包含有PKV全长的质粒,该研究为了解PKV在细胞中的复制机制,研制疫苗奠定了夯实的基础。为了实现上述目的,本发明采用的技术方案如下:1、一种猪嵴病毒全长cDNA的构建方法,其特征在于,所述方法包括如下步骤:S1利用Primerpremier5软件对拼接后CH441株全基因组序列和pBlueScriptKS(Ⅱ)载体进行序列分析,选择了载体和全基因组中都存在的单一酶切位点EcoRⅠ和NotⅠ,在全基因组中单一存在的MefⅠ及载体上不存在但通过引物在序列3’末端引入的PacⅠ酶切位点以及虽然全基因组序列上不止一个但可以通过调整顺序插入的SacⅠ和ClaⅠ酶切位点;利用这些酶切位点将全基因组连接在同一载体上;S2片段P1与pBlueScriptKS(Ⅱ)载体连接:将已克隆到pMD19-TSimpleVector载体上测序正确的pMD19-T-P1质粒用SacⅠ和EcoRⅠ限制性内切酶双酶切,反应体系为:10×NEBbuffer5μL;SacⅠ和EcoRⅠ限制性内切酶各2μL;pMD19-T-P1质粒10μL;去离子水31μL,混匀后37℃作用2h,回收后的片段与用相同的限制性内切酶酶切的pBlueScriptKS(Ⅱ)纯化回收产物用T4DNA连接酶进行连接,10μL反应体系如下:2×T4DNA连接酶1μL,T4DNA连接酶buffer1μL,载体1μL,P1片段7μL,混匀并16℃连接过夜后进行转化;次日挑取单菌落培养后,选出双酶切鉴定呈阳性的质粒送测序;将测序结果与已获得的PKVCH441株对应序列进行比对分析,测序正确的质粒命名为pBKS-P1;S3P2片段与pBKS-P1的连接:测序正确的pBKS-P1用EcoRⅠ和ClaⅠ限制性内切酶双酶切处理。回收后的片段与用相同方法处理的pMD19-T-P2回收产物用T4DNA连接酶进行连接,连接过夜后进行转化。挑选单菌落培养后提取质粒,选出双酶切鉴定呈阳性的质粒送测序;测序正确的质粒命名为pBKS-P1-P2;S43’片段与pBKS-P1-P2的连接:测序正确的pBKS-P1-P2用MefⅠ和ClaⅠ限制性内切酶双酶切处理。目的片段回收后与用相同方法处理的pMD19-T-3’回收产物用T4DNA连接酶连接过夜后进行转化;挑选单菌落培养后提取质粒,选出双酶切鉴定呈阳性的质粒送测序;将测序结果与已测得的PKVCH441株对应序列进行比对分析,测序正确者命名为pBKS-P1-P2-3’;S5P3片段与pBKS-P1-P2-3’的连接:测序正确的pBKS-P1-P2-3’用MefⅠ和NotⅠ限制性内切酶双酶切处理。回收后的片段与用相同方法处理的pMD19-T-P3回收产物连接过夜后进行转化;挑选单菌落培养适当时间后提取质粒,选出双酶切鉴定呈阳性的质粒送测序;测序结果与已测得的PKVCH441株对应序列进行比对分析,测序正确的质粒命名为pBKS-P1-P2-3’-P3;S6将测序合适的pBKS-P1-P2-3’-P3质粒用PacⅠ限制性内切酶单酶切,酶切反应在50μL体系中进行:10×NEBbuffer5μL;PacⅠ5μL;pBKS-P1-P2-3’-P3质粒10μL;去离子水30μL,混匀后37℃作用2h,然后进行纯化回收;S7回收的片段进行体外转录。所述纯化回收的步骤为:(1)将PCR产物转移到新的1.5mL离心管中,加入5倍体积的BufferCP;(2)将上述混合液混匀后转移到试剂盒提供的吸附柱中,10000g离心1min,弃滤液;(3)向吸附柱中加入700μLDNAWashBuffer,10000g离心1min,弃滤液;(4)重复步骤(3)一次;(5)全速离心2min;(6)将吸附柱套入新的1.5mL离心管,向吸附柱中垂直加入30μL洗脱液,静止2min;(7)全速离心2min,收集DNA。所述体外转录的步骤为:(1)将混合液轻轻混匀,37℃作用2h~4h;(2)向混合液中加入1μLTURBODNase,混匀后37℃孵育15min;转录后的混合液用RNeasyMinEluteCleanupKit纯化回收,步骤如下:(1)用DEPC水调整混合液的体积到450μL。(2)在上述混合液中加入250μL无水乙醇,轻轻混匀。(3)将上述混合液转入2mL的RNeasyMinElute管中,8000g离心15s后弃取液体。(4)将上述RNeasyMinElute收集管转入新的2mL柱子中,加入500μLRPE后8000g离心15s后弃去液体。(5)在上述混合液中加入500μL80%无水乙醇,轻轻混匀,8000g离心2min后弃液体和收集管。(6)将RNeasyMinElute柱子转移进入2mL离心管中,全速离心5min,弃收集管。(7)将RNeasyMinElute柱子转移进入1.5mL离心管中,加入14μLDEPC水,全速离心1min后收集液体。(8)回收后的RNA于-80℃保存备用。本发明有益效果在于,本研究通过RNA病毒的经典研究方法-反向遗传技术,成功构建了PKVCH441株全长质粒,并进行了体外转录,为其进一步深入研究PKV夯实了坚定的基础。附图说明图1为本发明PKVCH441基因组酶切位点选择示意图;图2为本发明猪嵴病毒全长质粒构建示意图;图3为本发明PKVCH441全基因组各片段的扩增;图4为不同PKV全基因组核苷酸序列相似性分析;图5为不同PKV全基因组进化树分析;图6为本发明的猪嵴病毒CH441全长质粒酶切鉴定图;具体实施方式下面通过具体的实施方式进一步对本发明进行描述,需要说明的是,所涉及的实施方式/实施例只是便于描述本发明的技术方案,并不是对本发明的限制。关于病毒、载体与菌株猪嵴病毒株CH441和pBlueScriptKS(Ⅱ)载体由中国农业科学院兰州兽医研究所动物病毒分子生态学创新团队保存;大肠杆菌DH5а受体菌和pMD19-TSimpleVector均购自宝生物工程(大连)有限公司。关于主要试剂和仪器主要试剂:MEGAscriptKit购自invitrogen公司;PlasmidMiniKitⅠ、GelExtractionKitⅠ及Cycle-PureKit均购自Omega公司;MiniBESTUniversalRNAExtractionKit试剂盒、OneStepPrimeScriptRT-PCRKit试剂盒、DL2000DNAMarker和DL5000DNAMarker均购自宝生物工程(大连)有限公司;T4DNA连接酶及限制性内切酶均购自美国纽英伦生物技术(北京)有限公司。主要仪器:移液枪购自Eppendorf公司;离心机购自Sigma公司;PCR仪购自德国Eppendorf公司;核酸电泳系统、UniversalHoodII凝胶成像系统均购自美国Bio-Rad公司;制冰机购自日本三洋公司;恒温摇床购自上海智城公司;恒温金属浴购自天根生化技术(北京)有限公司。关于PKVCH441全基因组扩增引物设计由于PKVCH4415’端序列高度保守且实验室已通过5’Race完成了其序列测定工作,在此基础上,我们将CH441株猪嵴病毒全基因组分为4段进行全基因RT-PCR扩增。扩增引物设计如下:关于全基因组分段扩增及测序1、重组质粒的构建回收目的基因片段克隆到pMD19-TSimpleVector。混匀体系后在恒温金属浴上16℃连接过夜后转化连接产物。次日从平板上挑取几个单菌落,培养适当时间后提取质粒。酶切鉴定阳性后送华大基因公司测序。关于全基因组序列的拼接与分析测序鉴定正确后,将包含PKVCH441全序列的4个片段进行序列比对分析,分别选出4个片段的优势序列,将选出的序列用DNAStar软件进行拼接获得CH441全基因组序列,将后者与登录在GenBank中的PKV全基因组序列进行比对分析。关于限制性内切酶位点的选择如图1所示,利用Primerpremier5软件对拼接后CH441株全基因组序列和pBlueScriptKS(Ⅱ)载体进行序列分析,选择了载体和全基因组中都存在的单一酶切位点EcoRⅠ和NotⅠ,在全基因组中单一存在的MefⅠ及载体上不存在但通过引物在序列3’末端引入的PacⅠ酶切位点以及虽然全基因组序列上不止一个但可以通过调整顺序插入的SacⅠ和ClaⅠ酶切位点。利用这些酶切位点将全基因组连接在同一载体上。关于CH441全基因组分段如图2所示将已克隆到pMD19-TSimpleVector上的测序合适的质粒按合适的顺序和酶切位点酶切并连接到pBlueScriptKS(Ⅱ)载体上,构建PKVCH441全长质粒。片段P1与pBlueScriptKS(Ⅱ)载体连接:将已克隆到pMD19-TSimpleVector载体上测序正确的pMD19-T-P1质粒用SacⅠ和EcoRⅠ限制性内切酶双酶切,反应体系为:10×NEBbuffer5μL;SacⅠ和EcoRⅠ限制性内切酶各2μL;pMD19-T-P1质粒10μL;去离子水31μL,混匀后37℃作用2h,回收后的片段与用相同的限制性内切酶酶切的pBlueScriptKS(Ⅱ)纯化回收产物用T4DNA连接酶进行连接,10μL反应体系如下:2×T4DNA连接酶1μL,T4DNA连接酶buffer1μL,载体1μL,P1片段7μL,混匀并16℃连接过夜后进行转化。次日挑取单菌落培养后用PlasmidMiniKitI提取质粒,选出双酶切鉴定呈阳性的质粒送测序。将测序结果与已获得的PKVCH441株对应序列进行比对分析,测序正确的质粒命名为pBKS-P1。P2片段与pBKS-P1的连接:测序正确的pBKS-P1用EcoRⅠ和ClaⅠ限制性内切酶双酶切处理。回收后的片段与用相同方法处理的pMD19-T-P2回收产物用T4DNA连接酶进行连接,连接过夜后进行转化。挑选单菌落培养后提取质粒,选出双酶切鉴定呈阳性的质粒送测序。测序正确的质粒命名为pBKS-P1-P2。3’片段与pBKS-P1-P2的连接:测序正确的pBKS-P1-P2用MefⅠ和ClaⅠ限制性内切酶双酶切处理。目的片段回收后与用相同方法处理的pMD19-T-3’回收产物用T4DNA连接酶连接过夜后进行转化。挑选单菌落培养后提取质粒,选出双酶切鉴定呈阳性的质粒送测序。将测序结果与已测得的PKVCH441株对应序列进行比对分析,测序正确者命名为pBKS-P1-P2-3’。P3片段与pBKS-P1-P2-3’的连接:测序正确的pBKS-P1-P2-3’用MefⅠ和NotⅠ限制性内切酶双酶切处理。回收后的片段与用相同方法处理的pMD19-T-P3回收产物连接过夜后进行转化。挑选单菌落培养适当时间后提取质粒,选出双酶切鉴定呈阳性的质粒送测序。测序结果与已测得的PKVCH441株对应序列进行比对分析,测序正确的质粒命名为pBKS-P1-P2-3’-P3。关于PKVCH441的体外转录将测序合适的pBKS-P1-P2-3’-P3质粒用PacⅠ限制性内切酶单酶切,酶切反应在50μL体系中进行:10×NEBbuffer5μL;PacⅠ5μL;pBKS-P1-P2-3’-P3质粒10μL;去离子水30μL,混匀后37℃作用2h,然后根据Cycle-PureKit说明书进行纯化回收。纯化回收步骤如下:(1)将PCR产物转移到新的1.5mL离心管中,加入5倍体积的BufferCP;(2)将上述混合液混匀后转移到试剂盒提供的吸附柱中,10000g离心1min,弃滤液;(3)向吸附柱中加入700μLDNAWashBuffer,10000g离心1min,弃滤液;(4)重复步骤(3)一次;(5)全速离心2min;(6)将吸附柱套入新的1.5mL离心管,向吸附柱中垂直加入30μL洗脱液,静止2min;(7)全速离心2min,收集DNA。回收的片段按MEGAscriptKitprocedure说明体外转录,体系如下:AmountComponentto20μLNuclease-freeWater2μLATPsolution2μLCTPsolution2μLGTPsolution2μLUTPsolution2μL10×ReactionBuffer0.1-1μgLineartemplateDNA+2μLEnzymeMix步骤为:(1)将上述混合液轻轻混匀,37℃作用2h~4h;(2)向上述混合液中加入1μLTURBODNase,混匀后37℃孵育15min;转录后的混合液用RNeasyMinEluteCleanupKit纯化回收,步骤如下:(1)用DEPC水调整混合液的体积到450μL。(2)在上述混合液中加入250μL无水乙醇,轻轻混匀。(3)将上述混合液转入2mL的RNeasyMinElute管中,8000g离心15s后弃取液体。(4)将上述RNeasyMinElute收集管转入新的2mL柱子中,加入500μLRPE后8000g离心15s后弃去液体。(5)在上述混合液中加入500μL80%无水乙醇,轻轻混匀,8000g离心2min后弃液体和收集管。(6)将RNeasyMinElute柱子转移进入2mL离心管中,全速离心5min,弃收集管。(7)将RNeasyMinElute柱子转移进入1.5mL离心管中,加入14μLDEPC水,全速离心1min后收集液体。(8)回收后的RNA于-80℃保存备用。关于全基因组各片段的扩增结果如图3所示,包含CH441全长的4个片段扩增结果。P1、P2、P3及3’大小分别为1265bp、3099bp、2673bp和1429bp。通过1.0%琼脂糖核酸凝胶分析各片段扩增结果与预期结果相符。关于CH441全基因组序列比对分析结果序列拼接结果显示,PKVCH441株全基因组全长为8148nt,A+T百分含量为47.83%,G+C百分比为52.17%。基因组成如下表:如图4、图5所示,利用MegAlign软件对NCBI中提交的部分PKV全基因组与PKVCH441株进行比对分析,结果显示,核苷酸相似度在到87.9%~90.8%之间,并且2B区域缺失90nt,这可能与自然选择和病原与宿主间的相互作用有关。关于构建全长cDNA双酶切鉴定图如图6所示,pBKS-P1双酶切片段分别为3000bp和1265bp;pBKS-P1-P2双酶切片段分别为4162bp和3000bp;pBKS-P1-P2-3’双酶切片段分别为7162bp和1429bp;pBKS-P1-P2-3’-P3双酶切片段分别为10162bp和2699bp;与预期结果一致,说明全长质粒构建成功。线性化的全长质粒体外转录后纯化的RNA浓度可达4000ng/μL反向遗传操作技术即利用现代生物学理论与方法,通过对核苷酸水平的操作来研究生物表型变化的一种手段,它与经典的从表型改变到基因功能研究的思路相反。目前采用的根据病毒的核苷酸序列,运用反向遗传操作技术来拯救RNA病毒的方法不仅可以克服体内RNA复制酶保真性低的缺点而且可为制备高免疫源性的低致病力毒株疫苗提供新的途径,同时能为研究RNA病毒的致病机制提供了一种有效手段。RACE技术即cDNA末端快速扩增技术又称锚定PCR,是由已知cDNA序列,扩增完整5’和3’末端的一种简便、有效的方法,包括3’race和5’race两种。同5’race相比,3’race更易成功且扩增片段长度更长。其基本原理为:利用mRNA的3’末端的多聚腺苷酸尾巴作为引物结合的位点,以通用接头引物的Oligo(dT)为引物反转录成cDNA。然后以反转录得到的cDNA为模板,特异引物GSP1和含有部分接头序列的通用引物UPM为上、下游引物进行PCR扩增。5’race第一步同3’race不同之处为:反转录达到cDNA5’末端时自动加上3~5个胞嘧啶残基,后者可以5’raceadapterprimer碱基配对,转换为含有SMART接头序列的cDNA为模板,然后以该cDNA为模板,含有部分接头序列的通用引物UPM和基因特异引物GSP2为上、下游引物,进行PCR扩增。随着PCR克隆技术的完善及RACE技术的不断改进和发展,RACE技术在基因克隆及基因表达研究中的作用将越来越显著。pBlueScriptKS(Ⅱ)是将T3、T7或SP6噬菌体启动子序列,组入噬菌体载体的多克隆位点区两侧,构建而成的体外转录载体。在体外系统中,加入合适的噬菌体RNA聚合酶就可使插入该载体的外源基因得到转录。由于RNA聚合酶的活性具有高度的专一性,它们只能识别噬菌体自己启动子的特定序列所以不会发生质粒基因的转录。其结构特点为:小分子量的共价、闭合、环状的基因组DNA,T3和T7噬菌体启动子分别位于多克隆位点的两侧,可定向启动外源基因的转录活性。该载体的特点为:含有氨苄青霉素抗性基因,可作为转化克隆的选择标记;拷贝数高;存在多克隆位点,可克隆达10kb的外源基因;可用组织化学显色反应法筛选噬菌体载体的重组子;含有质粒复制起点,即使在无辅助质粒存在的情况下,克隆的外源基因也可复制。因此,它不仅可以用于制备筛选cDNA文库或者基因组文库的放射性同位素标记的DNA分子探针;其体外转录合成的转录本还可以用来编码具有外源基因正确读码结构的外源蛋白。本研究通过RNA病毒的经典研究方法-反向遗传技术,成功构建了PKVCH441株全长质粒,并进行了体外转录,为其进一步深入研究PKV夯实了坚定的基础。对于本领域的技术人员来说,可根据以上描述的技术方案以及构思,做出其它各种相应的改变以及变形,而所有的这些改变以及变形都应该属于本发明权利要求的保护范围之内。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1