一种产碱性果胶酶的重组菌及其应用的制作方法
本发明涉及一种产碱性果胶酶的重组菌及其应用,属于基因工程技术领域。
背景技术:
果胶酶是一种复合酶,能够将果胶聚合物分解成不饱和寡聚半乳糖醛酸。该酶分布广泛,在部分寄生线虫、植物和微生物内都有发现。果胶酶应用广泛,已有40多年的工业应用史。根据最适反应pH的不同将果胶酶分为酸性果胶酶和碱性果胶酶(Alkaline Polygalacturonate Lyase,PGL)。其中酸性果胶酶主要应用于澄清果汁果酒,提取果蔬汁,果实脱皮等方面。PGL应用主要应用于纺织、食品、造纸行业和环境领域。应用酶法作用上述领域相关反应具有环保、节约原料耗材和反应条件温和等优点。然而目前对PGL进行分子改造研究较少,进行商品化的PGL也很少。
目前表达碱性果胶酶的宿主主要是毕赤酵母、枯草芽孢杆菌和大肠杆菌。虽然通过发酵优化等手段可以有效提高碱性果胶酶的产量,但当达到一定限度碱性果胶酶的产量不能进一步提高,限制了碱性果胶酶的工业化生产,因此需从源头解决限制碱性果胶酶表达的因素。
毕赤酵母宿主虽然具有表达蛋白易于纯化等优点,然而目前的碱性果胶酶在毕赤酵母中表达时,存在表达量不高或者是外源基因原始核苷酸序列并不十分适合于毕赤酵母宿主的问题,从而限制了碱性果胶酶在毕赤酵母中的高效表达,酵母真核表达系统中存在的糖基化现象对碱性果胶酶的高效表达及性质存在极大影响。
因此,有必要对碱性果胶酶糖基化位点的处理,进一步提高碱性果胶酶生产菌株的生产能力以更适应工业化的需要。
技术实现要素:
为了解决上述问题,本发明提供了一种糖基化位点删除的碱性果胶酶,以及表达该碱性果胶酶的毕赤酵母基因工程菌。
本发明的一个目的是提供一种产碱性果胶酶的基因工程菌,是以毕赤酵母为宿主,以pPIC9K为表达载体,表达序列如SEQ ID NO.1所示的碱性果胶酶基因,并以pGAPZA为载体,共表达ERO1和UBC1基因。
在本发明的一种实施方式中,所述ERO1基因GenBank登录号为XM_002489600.1,所述UBC1基因GenBank登录号为XM_002493814.1。
本发明的第二个目的是提供构建所述基因工程菌的方法,以毕赤酵母为宿主,以pPIC9K为表达载体,表达序列如SEQ ID NO.1所示的碱性果胶酶基因;以pGAPZA为载体,共表达ERO1和UBC1基因。
在本发明的一种实施方式中,所述方法步骤如下:
(1)将核苷酸序列如SEQ ID NO.1所示的碱性果胶酶基因与表达载体pPIC9K连接,转化至毕赤酵母中,获得Pichia pastoris GS115/N185Q;
(2)合成ERO1及UBC1基因;
(3)将步骤(2)获得的ERO1及UBC1基因分别连接到载体pGAPZA上,得到重组质粒pGAPZA-ERO1,pGAPZA-UBC1;
(4)将步骤(3)获得的重组质粒pGAPZA-ERO1、pGAPZA-UBC1,根据BglⅡ与BamHⅠ为同尾酶效应构建双基因组合共表达载体pGAPZA-ERO1-UBC1;
(5)将步骤(4)获得的重组质粒pGAPZA-ERO1-UBC1转化重组表达碱性果胶酶的毕赤酵母中,获得Pichia pastoris GS115/N185Q-ERO1-UBC1。
本发明的第三个目的是提供一种生产碱性果胶酶的方法,是将所述的基因工程菌接种至BMGY培养基中,发酵生产碱性果胶酶。
在本发明的一种实施方式中,所述方法是将基因工程菌接种至BMGY培养基中培养16~24h,然后再转接至含有占培养基体积7-10%的甲醇的BMMY中,于22-28℃,200~220rpm培养,每24h添加终浓度为10-20mL/L发酵液的甲醇。
在本发明的一种实施方式中,所述BMGY培养基每升含:蛋白胨20g,酵母粉10g,甘油40g,YNB 13.4g,pH6.0的0.1mol的磷酸盐缓冲液。
在本发明的一种实施方式中,所述BMMY培养基每升含:蛋白胨20g,酵母粉10g,70-100mL甲醇,YNB 13.4g,pH6.0的0.1mol的磷酸盐缓冲液。
在本发明的一种实施方式中,所述方法是将基因工程菌接种至BMGY培养基中,于30℃、220rpm条件下培养24h,然后再转接至含有占培养基体积7-10%的甲醇的BMMY中,于22-28℃、200~220rpm培养,每24h添加终浓度为10-20mL/L发酵液的甲醇。
在本发明的一种实施方式中,所述方法是所述基因工程菌在液体培养基中活化,将活化后的菌液以10~15mL菌液/100mL培养基的接种量接种至装液量500~1000mL发酵培养基的3L发酵罐中,初始搅拌转速为500~550r/min,通气量为1.5~2vvm,控制pH5.5-6.0,生长期培养温度为28-30℃;当甘油耗尽溶氧反弹时以指数流加方式补加甘油,待甘油再次耗尽溶氧反弹时,饥饿培养1~2h,开始流加诱导培养基,同时将温度降低至22-28℃,搅拌转速升高至900-1000r/min,所述诱导培养基是含有12ml/L微量元素PTM1的甲醇。
在本发明的一种实施方式中,所述方法在流加诱导培养基的同时还流加500g/L的山梨醇,流加速度为3-4g/(h·L)。
在本发明的一种实施方式中,所述培养基含有磷酸20~26ml/L,CaSO40.93g/L,K2SO418.2g/L,MgSO4·7H2O 14.9g/L,KOH 4.13g/L,甘油40.0g/L,微量元素PTM14.35ml/L。
本发明的第四个目的是提供所述基因工程菌在食品、纺织、环境、造纸方面的应用。
本发明的有益效果:本发明的基因工程菌株Pichia pastoris GS115/N185Q-ERO1-UBC1在摇瓶发酵时酶活比未表达分子伴侣的菌株Pichia pastoris GS115/N185Q提高了54.2%,且相比单独共表达单个分子伴侣的重组菌株Pichia pastoris GS115/N185Q-ERO1及Pichia pastoris GS115/N185Q-UBC1也分别提高了16.8%及23.4%,在3L发酵罐培养时,在甲醇单独诱导条件时Pichia pastoris GS115/N185Q-ERO1-UBC1最大酶活达到3885.04U/mL,相比于未共表达分子伴侣的重组菌株GS115/N185Q的2411.57U/mL提高了61.1%;当使用山梨醇与甲醇混合流加策略时,GS115/N185Q-ERO1-UBC1最大酶活达到5485.68U/mL,为目前文献报道中PGL的最高产量,实现了碱性果胶酶高效表达。本发明的碱性果胶酶可在碱性条件下催化通过反式消去作用聚半乳糖醛酸的α-1,4糖苷键裂解,广泛应用于食品、纺织和造纸等行业。
说明书附图
图1为克隆表达质粒示意图;
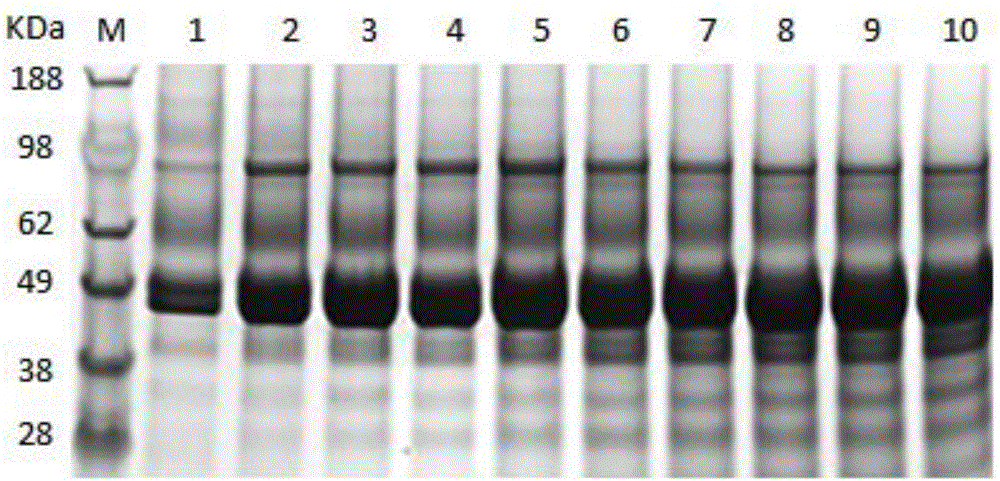
图2为分批补料发酵过程中发酵上清SDS-PAGE电泳分析;泳道1-10分别为诱导12h、24h、36h、48h、60h、72h、84h、96h、108h、120h发酵上清样品;
图3为共表达重组菌株摇瓶发酵性能;
图4为重组菌株在分批补料发酵过程发酵性能,a代表Pichia pastoris GS115/N185Q,b代表Pichia pastoris GS115/N185Q-ERO1-UBC1;
图5为重组菌株GS115/N185Q-ERO1-UBC1在分批补料发酵过程中在不同诱导策略下发酵性能,a代表甲醇诱导,b代表甲醇山梨醇混合流加诱导。
具体实施方式:
样品预处理:发酵液8000rpm离心10min,胞外PGL即包含于发酵上清液之中,取一定量做检测。
碱性果胶酶酶活测定:将发酵液8000rpm离心10min,胞外PGL即包含于发酵上清液之中,取一定量做检测。
PGL反应体系:含0.2%聚半乳糖醛酸(底物)的甘氨酸-NaOH缓冲液(0.2mol·L-1,0.44mmol·L-1的CaCl2,pH9.4)2mL,待测样品20μL,无活性的酶液为空白对照。
PGL反应条件为:将反应体系置于45℃下水浴15min,用3mL磷酸溶液(0.03mol·L-1)终止反应,在235nm处测定吸光度值。
单位酶活定义:单位时间裂解聚半乳糖醛酸产生1μmol的不饱和聚半乳糖醛酸所用的酶量。
培养基:
种子培养基YPD:胰蛋白胨20g/L、酵母粉10g/L、葡萄糖20g/L。
BMGY培养基(1L):蛋白胨20g,酵母粉10g,甘油40g,YNB13.4g,以pH6.0的0.1M的磷酸盐缓冲液调pH至pH6.0。
发酵培养基:85%磷酸26.7ml/L,CaSO40.93g/L,K2SO418.2g/L,MgSO4·7H2O 14.9g/L,KOH 4.13g/L,甘油40.0g/L,微量元素PTM14.35ml/L。
实施例1重组菌Pichia pastoris GS115/N185Q的构建
以基因K314Mopt(申请号为201610170070.0,公开日为2016年7月13日的专利申请中公开)为起始对照基因,通过点突变改造形成糖基化位点删除的碱性果胶酶基因PGL/N185Q,再设计引物,通过PCR的方法获得碱性果胶酶基因N185Q(序列如SEQ ID NO.1所示),将其克隆至表达载体pPIC9K上,获得重组质粒pPIC9K-N185Q,将重组载体转化Pichia pastoris GS115,经筛选鉴定获得重组菌株Pichia pastoris GS115-pPIC9K-N185Q。
引物如下:
PGL-F:GCTGAAGCTTACGTAGAATTCGCTGATTTGGGTCATCAAACACTTG
PGL-R:AAGGCGAATTAATTCGCGGCCGCTTAGTTCAATTTTCCAGCACCTGCT
采用电转化法将基因转入毕赤酵母细胞。具体步骤如下:挑取酵母受体菌的单菌落接种于25mL YPD液体培养基中,30℃摇床过夜;以5%接种量转接50mL YPD液体培养基,30℃摇床培养至OD600=1.3-1.5;4℃离心5000rpm,5min,弃上清;用50mL冰预冷无菌水将菌体重悬;4℃离心5000rpm,5min,弃上清;用25mL冰预冷无菌水将菌体重悬;4℃离心5000rpm,5min,弃上清;再用5mL 1mol·L-1的冰预冷的山梨醇洗涤1次,重悬,4℃,5000rpm离心5min,弃上清;加入适量体积1mol·L-1的冰预冷的山梨醇,重悬;分装至无菌EP管中,每管80ul,以备转化。将提取的共表达载体pPIC9K-PGL用酶Sal I线性化,在80μl酵母感受态细胞中加入线性化的质粒1-5μg冰上放置15分钟,迅速加入0.2cm电击杯中(冰预冷),1500v电击,迅速加入1mL冰预冷的山梨醇,涂MD平板,培养3-4天后挑取单克隆,测序验证获得重组菌Pichia pastoris GS115/N185Q。
实施例2Pichia pastoris GS115/N185Q-ERO1-UBC1的构建
提取毕赤酵母RNA,反转录为cDNA,以cDNA为模板,设计引物,通过PCR的方法获得ERO1及UBC1基因,将其克隆至表达载体pGAPZA上,获得重组质粒pGAPZA-ERO1及pGAPZA-UBC1,再根据BglⅡ与BamHⅠ为同尾酶效应构建双基因组合共表达载体pGAPZA-ERO1-UBC1(克隆表达质粒示意图见附图1),将重组载体pGAPZA-ERO1-UBC1转化Pichia pastoris GS115-pPIC9K-N185Q(实施例1中制备),经筛选鉴定获得共表达重组菌株Pichia pastoris GS115/N185Q-ERO1-UBC1。
引物如下:
毕赤酵母的转化采用电转化法。
具体步骤如下:挑取酵母受体菌的单菌落接种于25mLYPD液体培养基中,30℃摇床过夜;以5%接种量转接50mLYPD液体培养基,30℃摇床培养至OD=1.3-1.5;4℃离心,5000rpm,5min,弃上清;用50mL冰预冷无菌水将菌体重悬;4℃离心5000rpm,5min,弃上清;用25mL冰预冷无菌水将菌体重悬;4℃离心5000rpm,5min,弃上清;再用5mL 1mol/L的冰预冷的山梨醇洗涤1次,重悬,4℃,5000rpm离心5min,弃上清;加入适量体积1mol/L的冰预冷的山梨醇,重悬;分装至无菌EP管中,每管80μl,以备转化。将提取的共表达载体pGAPZA-X用酶AvrII线性化,在80μl酵母感受态细胞中加入用合适酶切位点线性化的质粒1-5μg冰上放置15分钟,迅速加入0.2cm电击杯中(冰预冷),1500v电击,迅速加入1ml冰预冷的山梨醇,涂含有200μg/mL Zeocin的YPDS平板,培养3-4天后挑取单克隆。
实施例3共表达基因工程菌株摇瓶发酵培养
培养方法:菌株在种子活化后接种到基本发酵培养基YPD,于30℃、220rpm条件下培养14h,转接至优化后的生长培养基BMGY培养基于30℃、220rpm条件下培养24h,再将菌株转入诱导培养基BMMY中22-28℃、220rpm每24h添加终浓度10~20mL/L的甲醇,诱导碱性果胶酶的表达。
选用碧云天SDS-PAGE凝胶电泳试剂盒配制12%分离胶和5%浓缩胶,具体操作方法见产品说明书。样品与5×上样缓冲液以体积比4:1混合,沸水浴10min,冷却后上样。电泳时,80V恒压电压,待指示剂进入分离胶后,电压调至150V,待指示剂至胶底时结束电泳。用考马斯亮蓝染色液对凝胶进行染色,染色1h后脱色(SDS-PAGE图谱见附图2)。
摇瓶诱导发酵96h时,重组菌Pichia pastoris GS11/N185Q-ERO1-UBC1的酶活为873.23U/mL,相比于共表达分子伴侣前的出发菌株Pichia pastoris GS115-pPIC9K-N185Q(酶活为566.40U/mL)及单独共表达单个分子伴侣的重组菌株Pichia pastoris GS115/N185Q-ERO1(酶活747.66U/mL)及Pichia pastoris GS115/N185Q-UBC1(酶活707.6U/mL),分别提高了54.2%、16.8%及23.4%(见附图3)。
实施例4 3L发酵罐发酵培养
按下述方法将Pichia pastoris GS115/N185Q和Pichia pastoris GS115/N185Q-ERO1-UBC1分别在相同条件下进行发酵:从固体培养基平板上挑取单菌落接种于YPD培养基(500mL三角瓶中装液量50mL)中30℃,220rpm培养24h作为种子液,然后以10%的接种量(10mL菌液接种至100mL培养基)接种于含800mL分批发酵培养基的3L发酵罐(美国NBS公司)中,初始搅拌转速为500r/min,通气量为2vvm,50%氨水及30%磷酸控制pH5.5,生长期培养温度为30℃,当甘油耗尽溶氧反弹时以指数流加方式补加质量浓度为500g/L的甘油(含12mL/L PTM1),流加速率按下式计算:F(t)为流加速率(L/h),X0为细胞密度(g/L),V0为初始体积(L),Sf代表补料液中甘油浓度(g/L),YX/S对底物细胞得率(g/L),μset为设定的比生长速率(h-1)。其中μset为0.176h-1,YX/S为0.435g/g,Sf为500g/L。
待甘油再次耗尽溶氧反弹时,饥饿培养1h,开始流加诱导培养基(100%甲醇含12mL/L PTM1),同时温度降低至22℃,搅拌转速升高至900r/min,诱导PGL表达。诱导培养基采用分阶段流加方式:0-8h流速2mL/h,8-90h流速9.6mL/h,>90h流速2mL/h。每隔12h取样一次,测定生物量,酶活,蛋白含量等参数。
实施例5 3L发酵罐发酵培养
具体实施方式同实施例4,其区别在于流加诱导培养基的同时还流加500g/L的山梨醇,流加速度为3-4g/(h·L)。
对实施例4和实施例4发酵结束后的发酵液中碱性果胶酶酶活进行检测,结果显示,在3L发酵罐对Pichia pastoris GS115/N185Q及Pichia pastoris GS115/N185Q-ERO1-UBC1进行分批补料发酵,以甲醇单独诱导时,Pichia pastoris GS115/N185Q-ERO1-UBC1最大酶活达到3885.04U/mL(重组菌分批补料发酵性能见图4),相比于未共表达分子伴侣的重组菌株GS115/N185Q的2411.57U/mL有明显提高,提高了61.1%;当使用山梨醇与甲醇混合流加策略时,GS115/N185Q-ERO1-UBC1最大酶活达到5485.68U/mL(图5),为现有技术报道中PGL的最高产量,实现了碱性果胶酶高效表达。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!