喹唑啉类化合物及其制备方法和用途与流程
本发明涉及医药
技术领域:
,具体涉及一种喹唑啉类化合物及其制备方法和用途。
背景技术:
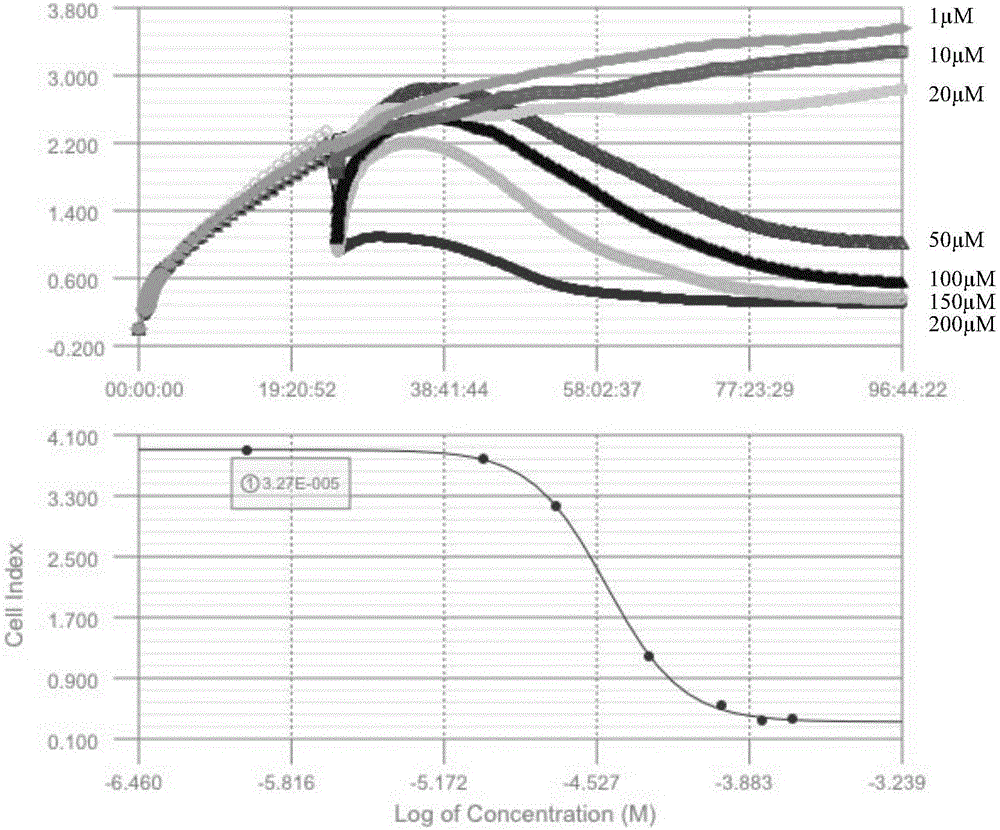
:黏着斑激酶(Focaladhesionkinase,FAK)是一种120KDa的非受体型酪氨酸激酶,因最早在细胞黏着斑中被发现而得名,在整合素介导的细胞信号通路中扮演着中枢的角色。FAK存在激活与非激活两种构象,处于非激活状态的FAK在整合素的作用下,构象发生改变,使得酪氨酸残基Y397发生磷酸化,FAK转变为激活状态,激活后的FAK与Src家族蛋白结合形成复合物,共同激活下游复杂的信号通路,调节细胞的黏附、迁移、增殖、分化以及生物体内的血管新生。有研究表明,FAK在多种肿瘤细胞中有显著高表达,同时,FAK表达和活性的增高与肿瘤的转移和不良预后成正相关。因此,FAK是肿瘤治疗的一个潜在靶点。目前,已经进入临床研究的FAK抑制剂主要有辉瑞公司的PF-562271,Poniard制药公司的PND-1186以及Verastem公司的Defactinib(结构式如下),但仍未见有药物被批准上市用于肿瘤临床治疗。基于上述技术背景,有必要对结构新颖,活性显著的FAK小分子抑制剂进行开发研究,以满足临床肿瘤治疗的需求。技术实现要素:本发明要解决的技术问题是提供一类喹唑啉类化合物,该类化合物具有抑制黏着斑激酶(FAK)作用,可用于黏着斑激酶相关疾病的治疗,尤其是用于黏着斑激酶高表达肿瘤的治疗。为了解决上述技术问题,本发明通过如下技术方案实现:在本发明的一个方面,提供了一种式(I)化合物或其可药用盐,其中,L为0或1;Z为苯基、取代的苯基、五元及六元内酰胺环并苯基、烯丙酰基、或N,N-二甲基氨基-2-丁烯酰基,当Z为取代的苯基时,该苯基上的取代基选自-OCH3、-NO2、-CN、-F、-Cl、-(CH2)mCH3、-(CH2)mOH、-(CH2)mNH2、-(CH2)mNHCOCH3、吗啉基、哌嗪基或4-甲基哌嗪基,m为0-3的整数;X和Y是相同或不相同的原子,选自C或N原子;R1、R2、R3、R4和R5是相同或不相同的取代基,选自:-H、-(CH2)nCH3、-O(CH2)nCH3、-NH(CH2)nCH3、-N(CH3)SO2(CH2)nCH3、-NHSO2(CH2)nCH3、-NHCO(CH2)nCH3、-CONH(CH2)nCH3或-SO2(CH2)nCH3,n为0-2的整数;R6位于所在苯环的任意位置,其包括-H、-CN、-OH、-Cl、-F、-CH3、-CH2CH3或-OCH3。优选的,所述式(I)化合物包括下述结构的化合物:所述盐为式(I)化合物与酸形成的加成盐,所述酸包括盐酸、氢溴酸、硫酸、柠檬酸、酒石酸、乳酸、丙酮酸、乙酸、马来酸、甲磺酸、或对甲苯磺酸。在本发明的另一方面,还提供了一种药物组合物,该组合物包含安全有效量的上述化合物或其可药用盐及药学上可接受的载体。上述可接受的载体是无毒的、能辅助施用并且对化合物的治疗效果没有不利影响。此类载体可以是本领域的技术人员通常能得到的任何固体赋形剂、液体赋形剂、半固体赋形剂或者在气雾剂组合物中的气体赋形剂。固体药物赋形剂包括淀粉、纤维素、滑石、葡萄糖、乳糖、蔗糖、明胶、麦芽、稻米、面粉、白垩、硅胶、硬脂酸镁、硬脂酸钠、甘油硬脂酰酯、氯化钠、无水脱脂乳等。液体和半固体赋形剂可以选自甘油、丙二醇、水、乙醇和各种油,包括那些源于石油、动物、植物或人工合成的油,例如,花生油、豆油、矿物油、芝麻油等、优选的液体载体,特别是用于可注射溶液的,包括水、盐水、葡萄糖水溶液和甘醇。另外还可以在组合物中加入其它辅剂如香味剂、甜味剂等。本发明的化合物以治疗上的有效量施用,其施用方式可以是口服、全身施用(例如,透过皮肤的、鼻吸入的或者用栓剂)或肠胃外施用(例如,肌肉内、静脉内或皮下)。优选的施用方式是口服,它可根据疾病程度调节。本发明的化合物的实际施用量(即活性组分)依赖于许多因素,如待治疗疾病的严重性、治疗对象的年龄和相对健康程度、所使用的化合物的效能、施用途径和形式,以及其他因素。本发明药物组合物的各种剂型可以按照药学领域的常规方法制备。例如使该化合物与一种或者多种载体混合,然后将其制成所需的剂型,如片剂、药丸、胶囊、半固体、粉末、缓释剂型、溶液、混悬液、配剂、气雾剂等等。在本发明的另一方面,还提供了上述化合物的制备方法,包括以下步骤:第一步4位取代氨基的引入即中间体化合物式(II)的制备,由原料化合物A与式(III)所示胺试剂在适当溶剂中经催化反应制得,具体可通过以下两种方法获得:方法1,原料化合物A与1-1.5当量式(III)胺试剂在适当溶剂中,在碱性试剂的催化下,通过亲核取代反应获得;或者方法2,原料化合物A与1-1.5当量式(III)胺试剂在适当溶剂中,在钯试剂与含磷配体的催化下,通过偶联反应获得;其中,原料化合物A选自购买或者自制的5-取代-2,4-二氯吡咯并[2,3-d]嘧啶,其取代基R6位于所在苯环的任意位置,包括-H、-CN、-OH、-Cl、-F、-CH3、-CH2CH3或-OCH3;式(III)胺试剂为取代的芳胺或者取代的芳基甲胺,其中,X和Y是C或者N;R1、R2、R3、R4和R5是相同或不相同的取代基,选自-H、-(CH2)nCH3、-O(CH2)nCH3、-NH(CH2)nCH3、-N(CH3)SO2(CH2)nCH3、-NHSO2(CH2)nCH3、-NHCO(CH2)nCH3、-CONH(CH2)nCH3或-SO2(CH2)nCH3,n为0-2的整数;溶剂选自乙醇、异丙醇、正丁醇、叔丁醇、四氢呋喃、二氧六环、或者N,N-二甲基甲酰胺等;碱性试剂选自吡啶、三乙胺、N,N-二异丙基乙胺、碳酸钾、或碳酸铯等;钯试剂选自醋酸钯、氯化钯、三(二亚苄基丙酮)二钯、或四(三苯基膦)钯等;含磷配体选自Xantphos、Xphos、或BINAP等。第二步2位取代氨基的引入即式(II)中间体化合物与Z-NH2胺试剂在适当溶剂中,经催化反应制得式(I)所示的喹唑啉类化合物,具体可通过以下两种方法获得:方法1,上述第一步反应产物即式(II)中间体化合物与1-1.5当量Z-NH2胺试剂在适当溶剂中,在碱性试剂的催化下,通过亲核取代反应获得;或者方法2,式(II)中间体化合物与1-1.5当量Z-NH2胺试剂在适当溶剂中,在钯试剂与含磷配体催化下,通过偶联反应获得;其中,Z-NH2胺试剂中Z为苯基、取代的苯基、五元及六元内酰胺环并苯基、烯丙酰基、或N,N-二甲基氨基-2-丁烯酰基;当Z为取代的苯基时,该苯基上的取代基选自-OCH3、-NO2、-CN、-F、-Cl、-(CH2)mCH3、-(CH2)mOH、-(CH2)mNH2、-(CH2)mNHCOCH3、吗啉基、哌嗪基或4-甲基哌嗪基,m为0-3的整数;溶剂选自乙醇、异丙醇、正丁醇、叔丁醇、四氢呋喃、二氧六环、或者N,N-二甲基甲酰胺等;碱性试剂选自吡啶、三乙胺、N,N-二异丙基乙胺、碳酸钾、或碳酸铯等;钯试剂选自醋酸钯、氯化钯、三(二亚苄基丙酮)二钯、或四(三苯基膦)钯等;含磷配体选自Xantphos、Xphos、或BINAP等。在本发明的另一方面,还提供了一种式(II)所示的药物中间体,其中,L为0或1;X和Y是相同或不相同的原子,选自C或N原子;R1、R2、R3、R4和R5是相同或不相同的取代基,选自:-H、-(CH2)nCH3、-O(CH2)nCH3、-NH(CH2)nCH3、-N(CH3)SO2(CH2)nCH3、-NHSO2(CH2)nCH3、-NHCO(CH2)nCH3、-CONH(CH2)nCH3或-SO2(CH2)nCH3,n为0-2的整数;R6位于所在苯环的任意位置,其包括-H、-CN、-OH、-Cl、-F、-CH3、-CH2CH3或-OCH3。在本发明的另一方面,还提供了上述化合物或其可药用盐在制备治疗黏着斑激酶相关疾病的药物中的应用。本发明化合物具有明显的黏着斑激酶(FAK)抑制活性,可用于黏着斑激酶相关疾病的治疗。在本发明的另一方面,还提供了上述化合物或其可药用盐在制备治疗癌症的药物中的应用。由于黏着斑激酶(FAK)是潜在的抗肿瘤药物靶点,而本发明的化合物具有显著的黏着斑激酶(FAK)抑制活性,通过实验证实这些化合物对FAK增高表达的各种癌细胞增殖具有抑制作用,因此本发明化合物适用于治疗各种癌症。本发明的喹唑啉类化合物,能够抑制FAK激酶的生物活性,能有效抑制肿瘤细胞生长,可用于黏着斑激酶(FAK)相关疾病的治疗,尤其适用于对FAK增高表达的癌症的治疗,应用前景非常广阔。附图说明下面结合附图和具体实施方式对本发明作进一步详细的说明。图1是本发明实施例1化合物抑制癌细胞生长的曲线图;图2是本发明实施例5化合物抑制癌细胞生长的曲线图;图3是本发明实施例7化合物抑制癌细胞生长的曲线图;图4是本发明实施例8化合物抑制癌细胞生长的曲线图。具体实施方式实施例1化合物1的合成297mg(1.28mmol)化合物A-1、275mg(1.28mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.356mL(2.56mmol)三乙胺溶于15mL二氯甲烷,室温搅拌。4h后停止反应,减压蒸去溶剂,所得粗品用乙醚洗涤,得到白色固体B-1,收率43.0%。100mg(0.24mmol)化合物B-1及36mg(0.24mmol)5-氨基吲哚啉-2-酮溶于3mL异丙醇,微波100℃反应1h。反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡黄色固体,即为化合物1,收率61.9%。m.p.183-190℃.1HNMR(DMSO-d6,400MHz)δ=3.03(s,3H),3.10(s,3H),3.27(s,2H),4.89(s,2H),6.66(brs,1H),7.05(brs,1H),7.16(s,1H),7.39(dd,J=7.52Hz,J=4.76Hz,1H),7.49-7.55(m,2H),7.73-7.79(m,2H),8.47(d,J=4.16Hz,1H),9.33(brs,1H),10.23(brs,1H),10.40(s,1H)ppm.ESI-MSm/z523.8[M+H]+.实施例2化合物1b的合成297mg(1.28mmol)化合物A-2、275mg(1.28mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.356mL(2.56mmol)三乙胺溶于15mL二氯甲烷,室温搅拌。反应4h后停止,减压蒸去溶剂,所得粗品用乙醚洗涤,得到米白色固体B-2(540mg),收率99.0%。100mg(0.24mmol)化合物B-2及36mg(0.24mmol)5-氨基吲哚啉-2-酮溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡绿色固体62mg,即为化合物1b,收率48.8%。m.p.209-213℃.1HNMR(DMSO-d6,400MHz)δ=3.06(s,3H),3.11(s,3H),3.35(s,2H),4.84(brs,2H),6.70(brs,1H),7.11(d,J=5.44Hz,1H),7.22(brs,1H),7.42(t,J=5.52Hz,1H),7.60(d,J=9.24Hz,1H),7.84-7.86(m,2H),8.49(m,2H),10.01(brs,1H),10.26(brs,1H),10.42(s,1H)ppm.ESI-MSm/z523.8[M+H]+.实施例3化合物1c的合成297mg(1.28mmol)化合物A-3、275mg(1.28mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.356mL(2.56mmol)三乙胺溶于15mL二氯甲烷,室温搅拌。反应4h后停止,减压蒸去溶剂,所得粗品用乙醚洗涤,得到白色固体B-3,收率70.3%。100mg(0.24mmol)化合物B-3及36mg(0.24mmol)5-氨基吲哚啉-2-酮溶于3mL异丙醇,微波100℃反应1h。反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡黄色固体,即为化合物1c,收率50.5%。m.p.>250℃.1HNMR(DMSO-d6,400MHz)δ=3.05(s,3H),3.11(s,3H),4.84(s,2H),6.70(s,1H),7.10(s,1H),7.21(s,1H),7.41(t,J=5.16Hz,1H),7.53(d,J=8.76Hz,1H),7.67(s,1H),7.83(s,1H),8.40(d,J=8.80Hz,1H),8.48(d,J=4.42Hz,1H),10.19(s,1H),10.38(s,1H),10.44(s,1H)ppm.ESI-MSm/z523.8[M+H]+.实施例4化合物1d的合成297mg(1.28mmol)化合物A-4、275mg(1.28mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.356mL(2.56mmol)三乙胺溶于15mL二氯甲烷,室温搅拌。反应4h后停止,减压蒸去溶剂,所得粗品用乙醚洗涤,得到白色固体B-4,收率68.9%。100mg(0.24mmol)化合物B-4及36mg(0.24mmol)5-氨基吲哚啉-2-酮溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡绿色固体,即为化合物1d,收率70.8%。m.p.231-236℃.1HNMR(DMSO-d6,400MHz)δ=3.03(s,3H),3.11(s,3H),3.28(s,2H),4.87(d,J=4.96Hz,2H),6.61(d,J=8.12Hz,1H),7.11(brs,1H),7.23(brs,1H),7.37-7.45(m,2H),7.82(d,J=7.68Hz,1H),8.00(d,J=6.72Hz,1H),8.33(d,J=7.24Hz,1H),8.48(d,J=4.08Hz,1H),10.36(s,2H),11.18(brs,1H),11.97(brs,1H)ppm.ESI-MSm/z523.8[M+H]+.实施例5化合物2的合成300mg(1.38mmol)化合物A-5、297mg(1.38mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.4mL三乙胺溶于15mL二氯甲烷,室温搅拌。反应4h后停止,减压蒸去溶剂,所得粗品用乙醚洗涤,得到米白色固体B-5,收率49.0%。100mg(0.25mmol)化合物B-5及37mg(0.25mmol)5-氨基吲哚-2-酮溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡绿色固体,即为化合物2,收率52.8%。m.p.187-190℃.1HNMR(DMSO-d6,400MHz)δ=3.03(s,3H),3.10(s,3H),3.27(s,2H),4.89(s,2H),6.66(brs,1H),7.05(brs,1H),7.16(s,1H),7.39(dd,J=7.52Hz,J=4.76Hz,1H),7.49-7.55(m,2H),7.73-7.79(m,2H),8.47(d,J=4.16Hz,1H),9.33(brs,1H),10.23(brs,1H),10.40(s,1H)ppm.ESI-MSm/z508.58[M+H]+.实施例6化合物3的合成300mg(1.34mmol)化合物A-6、288mg(1.34mmol)化合物N-(3-(氨甲基)吡啶-2-基)-N-甲基甲磺酰胺以及0.5mL三乙胺溶于15mL二氯甲烷,室温搅拌。反应4h后停止,减压蒸去溶剂,所得粗品用乙醚洗涤,得到白色固体B-6,收率65.0%。100mg(0.25mmol)化合物1-13a及37mg(0.25mmol)5-氨基吲哚-2-酮溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡黄色固体,即为化合物3,收率63%。m.p.192-195℃.1HNMR(DMSO-d6,400MHz)δ=3.03(s,3H),3.10(s,3H),3.27(s,2H),4.89(s,2H),6.66(brs,1H),7.05(brs,1H),7.16(s,1H),7.39(dd,J=7.52Hz,J=4.76Hz,1H),7.49-7.55(m,2H),7.73-7.79(m,2H),8.47(d,J=4.16Hz,1H),9.33(brs,1H),10.23(brs,1H),10.40(s,1H)ppm.ESI-MSm/z515.5[M+H]+.实施例7化合物4的合成298mg(1.28mmol)化合物A-7、150mg(1.28mmol)化合物间乙炔基苯胺以及0.356mL(2.56mmol)三乙胺溶于15mL二氯甲烷,室温搅拌过夜,减压蒸去溶剂,所得粗品用乙醚洗涤,得到白色固体B-7,收率72%。100mg(0.32mmol)化合物B-7及30mg(0.42mmol)烯丙酰胺溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡绿色固体,即为化合物4,收率71%。1HNMR(DMSO-d6,400MHz)δ=10.67(1H,s),10.40(1H,s),8.88(1H,s),7.87(1H,d,J=9.39Hz),7.71(1H,dd,J1=8.61Hz,J2=3.91Hz),7.49(1H,dd,J1=7.82Hz,J2=4.69Hz),7.35(1H,dd,J1=7.83Hz,J2=3.91Hz),7.26(1H,dd,J1=8.22Hz,J2=4.31Hz),6.50~6.57(1H,m),6.32(1H,dd,J1=16.83Hz,J2=1.57Hz),5.82(1H,dd,J1=11.74Hz,J2=1.57Hz)ppm.ESI-MSm/z349.8[M+H]+.实施例8化合物5的合成300mg(1.38mmol)化合物A-8、236mg(1.38mmol)化合物间溴苯胺以及0.4mL三乙胺溶于15mL二氯甲烷,室温搅拌过夜后停止反应,减压蒸去溶剂,所得粗品用乙醚洗涤,得到米白色固体B-8,收率69.0%。100mg(0.34mmol)化合物B-8及30mg(0.42mmol)烯丙酰胺溶于3mL异丙醇,微波100℃反应1h。将反应液过滤,滤饼用少量异丙醇及乙醚洗涤,干燥得淡绿色固体,即为化合物5,收率58%。1HNMR(DMSO-d6,400MHz)δ=10.47(1H,s,-NH-),10.35(1H,s,-NH-),8.88(1H,d,J=9.00Hz,7-H),8.03(1H,s,2’-H),7.83(1H,dd,J1=9.00Hz,J2=1.95Hz,7-H),7.78(1H,d,J=7.04Hz,8-H),7.71(1H,d,J=8.61Hz,6’-H),7.35~7.39(2H,m,5’-H,4’-H),6.46~6.57(1H,m,-CH),6.32(1H,dd,J1=18.78Hz,J2=1.57Hz,CH2=CH),5.82(1H,dd,J1=12.13Hz,J2=1.57Hz,CH2=CH).实施例9化合物6的合成75mg(0.195mmol)实施例5步骤(1)所得固体(化合物B-5)、56mg(0.293mmol)4-(4-甲基哌嗪)苯胺、81mg(0.585mmol)碳酸钾、9mg(0.00975mmol)Pd2(dba)3及9.3mg(0.0195mmol)Xphos溶于4mL叔丁醇,氮气保护,外浴100℃搅拌。反应15h后停止,向反应液中加入水与乙酸乙酯进行萃取,有机相干燥后柱层析,洗脱剂极性为二氯甲烷:甲醇=15:1,得到玫红色固体12mg,即为化合物6。收率11.4%。m.p.188-192℃.1H-NMR(DMSO-d6,400MHz)δ=2.24(s,3H),2.50(s,4H),3.00(s,4H),3.17(s,3H),3.19(s,3H),4.80(d,J=5.08Hz,2H),6.68(s,1H),6.72(d,J=8.52Hz,2H),7.22(brs,1H),7.42-7.45(m,3H),7.84(d,J=7.64Hz,1H),8.41(s,2H),10.78(s,1H)ppm.ESI-MSm/z539.9[M+H]+.实施例10化合物7的合成75mg(0.195mmol)实施例5步骤(1)所得固体(化合物B-5)、36mg(0.390mmol)苯胺、81mg(0.585mmol)碳酸钾、9mg(0.00975mmol)Pd2(dba)3及9.3mg(0.0195mmol)Xphos溶于4mL叔丁醇,氮气保护,外浴100℃搅拌。反应15h后停止,向反应液中加入水与乙酸乙酯进行萃取,有机相干燥后柱层析,洗脱剂极性为二氯甲烷:甲醇=150:1,得到淡黄色固体29mg,即为化合物7。收率33.7%。m.p.209-213℃.1H-NMR(DMSO-d6,400MHz)δ=3.18(s,3H),3.20(s,3H),4.83(d,J=5.28Hz,2H),6.74(s,1H),6.78(t,J=7.24Hz,1H),7.09(t,J=7.48Hz,2H),7.31(brs,1H),7.41(t,J=4.76Hz,1H),7.62(d,J=7.72Hz,2H),7.85(d,J=7.28Hz,1H),8.43(s,1H),8.70(s,1H),10.88(s,1H)ppm.ESI-MSm/z441.9[M+H]+.实施例11黏着斑激酶(FAK)抑制活性实验将上述实施例1-10的化合物进行黏着斑激酶(FAK)抑制活性的检测。1.方法40mMTris(pH7.4)、10nMMgCl2、0.1mg/mLBSA、1mMDTT、10μMATP、240ngFAK、0.2mg/mLPoly(Glu,Tyr)以及待测浓度的化合物组成50μL的反应体系,化合物的浓度梯度为0.1nM、0.33nM、1nM、3.3nM、10nM、33nM、100nM、333nM、1μM。反应体系在30℃下反应120min后,向体系中加入50μLKinase-GloPlus检测剂,30℃反应10min,MD-SpectraMaxM5多功能酶标仪Luminecesce模式下检测发光,收集数据,依据公式酶活性%={(Lu无酶—Lu加药)/(Lu无酶—Lu有酶无药)}*100%计算得到各浓度下的酶活性,再用GraphpadPrism5软件处理,拟合得到IC50值。2.实验结果表1实施例1-8的化合物对FAK激酶的抑制活性编号FAKIC50(μM)实施例10.210实施例210实施例310实施例410实施例50.055实施例60.206实施例70.750实施例80.702实施例90.013实施例100.0325PF-5622710.0011由表1的数据可知,本发明上述实施例的化合物具有FAK激酶的抑制活性。实施例10肿瘤细胞生长抑制实验1.方法(1)消化细胞,计数,吸取2X105细胞(SMMC7721、YY-8103等),稀释至6mL;根据其生长特性按1×103/100μl/孔计算细胞总量,并充分稀释后,种入96孔板内。每组每天3复孔,一般按6天接种细胞;(2)待半小时左右,细胞基本贴壁后观察细胞状态和数目。用显色剂显色反应,以确定细胞实际的起始密度,作为生长相对零点。显色剂Cellcountingkit-8(CCK-8,Dojindo,Japan)的用法:每100μl培液加10μlCCK-8,37℃、5%CO2放置1h,在酶标仪上测定450nm处的吸光度;(3)按照1:10稀释配制待测化合物的培养基各200ul,阳性化合物:200ulDMEM(含10%FBS)+3ul阳性化合物(10mM);(4)在E-plate中每孔加入150ul含化合物的培养基或阳性对照化合物的培养基;取出E-plate,向每孔中加入300ul细胞悬液、混匀;(5)光镜下观察细胞形态,以确定细胞生长状态。利用美国Biosciences公司ACEARTCA细胞分析仪定时测量,每天记录一次生长状况;一般需要测5至7天;(6)根据测量结果,绘制实验结果曲线。2.实验结果上述实施例化合物的检测结果如下表2和图1-4所示:表2实施例化合物对癌细胞生长的抑制结果编号IC50(μM)实施例5(化合物2)32.7实施例7(化合物4)13.4实施例8(化合物5)15.5实施例9(化合物6)2.98PF-562271(阳性对照)10.1图1是实施例1化合物抑制癌细胞生长的曲线图,实验为单测试浓度,测试浓度为50uM,其中A为肝癌细胞SMMC7721的检测结果,B为肝癌细胞YY-8103的检测结果。由图1可知,实施例1化合物(化合物1)能抑制肝癌细胞的生长。图2是实施例5化合物抑制癌细胞生长的曲线图,实验为多浓度测试,测试浓度从上至下依次为:1,10,20,50,100,150,200uM七个浓度,IC50=32.7μM。由图2可知,实施例5化合物(化合物2)能抑制癌细胞的生长。图3是实施例7化合物抑制癌细胞生长的曲线图,实验为多浓度测试,测试浓度从上至下依次为:1,5,10,20,30,50,100uM七个浓度,IC50=13.4uM。由图3可知,实施例7化合物(化合物4)能抑制癌细胞的生长。图4是实施例8化合物抑制癌细胞生长的曲线图,实验为多浓度测试,测试浓度从上至下依次为:1,5,10,20,50,100uM六个浓度,IC50=15.5uM。由图4可知,实施例8化合物(化合物5)能抑制癌细胞的生长。由上述表2和图1-4可知,本发明的化合物通过抑制FAK酪氨酸激酶的活性,能够有效抑制人肝癌细胞等各种有FAK增高表达特征的癌细胞的生长,适用于此类癌症的治疗。以上所述实施例仅表达了本发明的实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1