一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法与流程
本发明属于生物技术领域,具体地说,涉及一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法。
背景技术:
截止目前,miRNA的鉴别及发现主要有三种方法:miRNA cDNA文库测序法,生物信息学预测法和正向遗传性筛选。
生物信息学已经发现了大量新miRNA,但该方法存在一个弊端:它是通过对已知miRNA序列或者前体序列规律的总结去鉴别新miRNA,因此导致预测结果精确度不高,而且容易出现假阳性和假阴性。正向遗传学筛选是分子生物学家最初使用的一种研究方法,它是通过生物个体或细胞基因组的自发突变或人工诱变产生的变异表型而鉴定候选miRNA。使用该方法最早鉴定发现了lin-4,let-7,后来通过这种方法又鉴别发现了果蝇中的miR-278、miR-14以及线虫中的lsy-6。可是,通过正向遗传学筛选鉴别候选miRNA的效率比较低;如果miRNAs只是单个碱基发生突变、缺失、错位或碱基的改变仅仅使表型发生微小的差异甚至没有变化,造成许多候选miRNAs的鉴别发生遗漏,因此该方法已经慢慢被淘汰。
miRNA cDNA文库测序法包括直接克隆法和高通量测序法,直接克隆法效率相对比较低,费时费力,具有明显的局限性。相对于克隆测序技术,高通量测序具有高效性、精确性以及快速性等优点,提取RNA接头连接反转录后只进行PCR扩增,直接上机测序即可,不需要克隆过程,再将测序结果进行生物信息学分析及靶基因预测等,从而获得新miRNA。目前,高通量测序技术已经广泛应用到各种植物及动物的miRNA研究当中。目前,现有技术中还没有一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法。
技术实现要素:
本发明的目的在于提供一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法,该方法利用高通量测序技术及生物信息学技术对萨能奶山羊胎儿成纤维细胞miRNA进行鉴定并作GO功能分析及KEGG通路分析。
其具体技术方案为:
一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法,包括以下步骤:
步骤1、萨能奶山羊胎儿成纤维细胞的获得:
选取5只奶山羊做同期发情,与最后一次配种30天B超检查妊娠状况,选成功妊娠并与40-45天通过手术法无菌取出胎儿,用含双抗PBS冲洗三遍,放入无血清培养基快速带回实验室。在细胞室内无菌除去头、四肢、内脏,剩余皮肤组织酒精冲洗三遍,PBS冲洗三遍,然后剪成1mm3小块贴在细胞培养皿进行培养,每三天换一次培养基,待细胞铺满90%时消化传代,并作生长曲线及核型分析;
步骤2、RNA提取:
取对数生长期的细胞用trizol法提取RNA,该实验用到的所有物品包括1000mL、200μL、10μL tip头、枪头盒、Enpendoff管必须用0.1%的DEPC水或无RNA酶水浸泡6h以上或直接过夜,包装好后高压灭菌20-30min,65℃烘干备用;用到的所有研钵、玻璃器皿、金属器械用锡箔纸包装好后放于180℃烘箱中烘烤8h以上,自然降温备用,提取的总RNA经微量分光光度计检查纯度及浓度,合适以后送华大基因进行高通量测序;
步骤3、文库构建及高通量测序
取上述总RNA经PAGE胶纯化、反转录,然后以HiSeq2000为测序平台进行高通量测序构建cDNA文库;
步骤4、数据处理
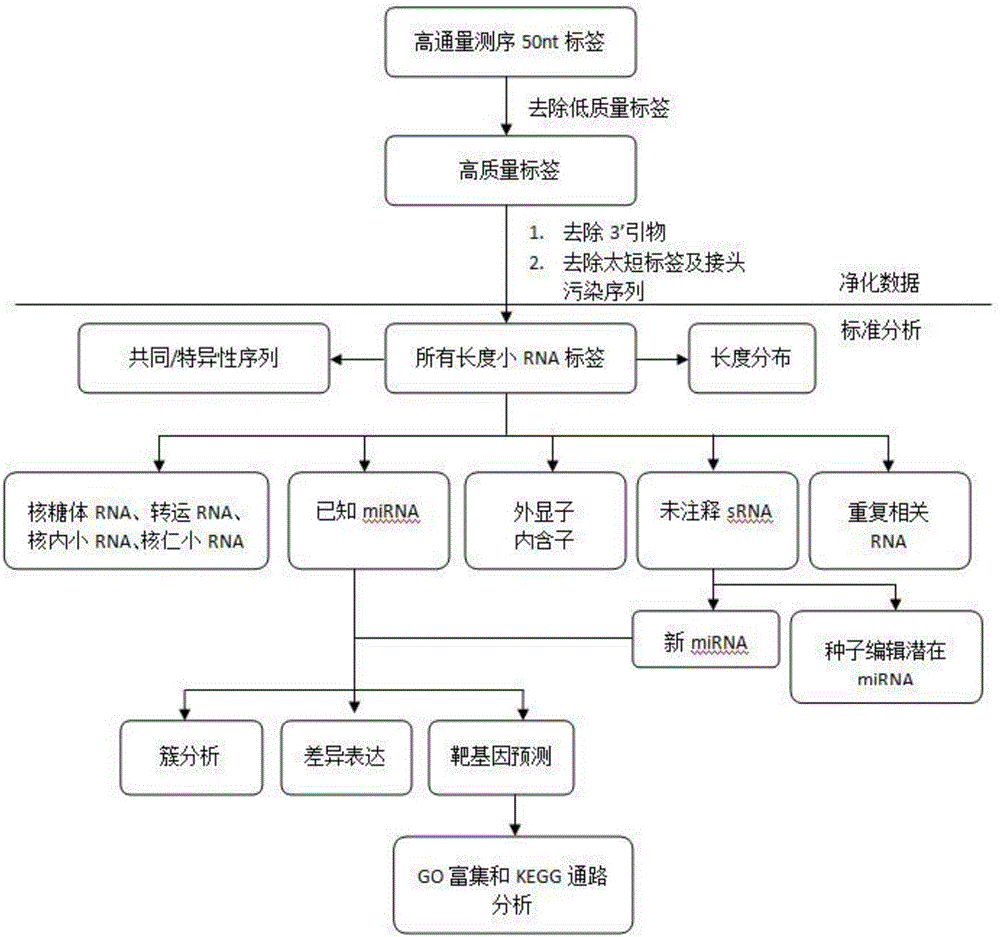
萨能奶山羊胎儿成纤维细胞样品在HiSeq2000平台测序后得到冗余数据,对该原始数据首先要进行去低质量序列,去接头序列以及去污染序列处理,获得干净序列即clean reads,然后再进行下一步生物信息学分析;
将获得的clean reads与绵羊全基因组数据库进行blast比对,用SOAPv1.11软件分析序列表达差异及分布情况,为了使每个unique sRNA有唯一的注释,我们采用如下优先规则:rRNAetc>known miRNA>repeat>exon>intron,由于rRNAetc是经Genbank数据库和Rfam数据库比对获得,实验规定两个数据库间的优先顺序为Genbank>Rfam,没有比对上任何注释信息的sRNA用unann表示。
为进一步分析候选miRNA二级结构,该实验用Mfold3.2软件]和RNAfold在线软件预测新miRNA颈环结构及最小自由能,用MIREAPv0.2软件再进一步深入分析每一个候选序列,参数设置如下:
(1)miRNA序列长度为18-26nt
(2)miRNA参考序列长度为20-24nt
(3)Drosha/Dicer酶切位点最少为3个
(4)miRNA在参考序列上最大拷贝数为20
(5)miRNA前体允许的最大自由能为200kcal/mol
(6)miRNA和miRNA*之间最大空格数为35
(7)miRNA和miRNA*之间最小配对数为14
(8)miRNAand miRNA*之间最大凸环为4。
进一步,步骤3中具体操作过程依据Small RNA Sample Preparation Kit(Illumina,USA)试剂盒说明书步骤进行,简单过程如下:
(1)首先通过15%聚丙酰胺凝胶电泳分离总RNA,切割片段为18-30nt部分,然后进行分离纯化、胶回收;
(2)通过T4RNA连接酶分别在3’端和5’端添加3’((PO4)-CTGTAGGCACCATCAA-(NH2)-(CH2)6)和5’(ATCGTAGGCACCUGAAA)接头序列
(3)将成功添加接头序列的RNA进行反转录成cDNA,然后25个循环进行PCR扩增。
(4)90bp左右的PCR扩增产物经PAGE胶纯化回收以后其产物在深圳华大科技HiSeq2000平台进行测序。
(5)测序片段去除低质量序列、接头序列、重复序列以及污染序列后进行信息生物学分析。
进一步,步骤4中进行去低质量序列,去接头序列以及去污染序列处理的过程具体如下:
(1)去除有5’接头序列的reads;
(2)去除没有3’接头序列的reads;
(3)去除测序质量较低的reads;
(4)去除没有插入片段的reads;
(5)去除含有polyA的reads;
(6)去除小于18核苷酸的片段。
与现有技术相比,本发明的有益效果:
本发明通过高通量测序,获得16395039 total_reads,取出冗余数据后获得clean_reads 16150181,其中Unique sRNAS 205857。生物信息学分析获得已知miRNA 44条,候选新miRNA247条。已知miRNA靶基因数量5401,靶基因位点数为6069;候选miRNA靶基因数量8401,靶基因位点数位10832。
附图说明
图1是:高通量测序准备流程图;
图2是:生物信息学分析流程图;
图3是:部分候选新miRNA前体二级结构。
具体实施方式
下面结合附图和具体实施方案对本发明的技术方案作进一步详细地说明。
一种萨能奶山羊胎儿成纤维细胞miRNA的筛选与鉴定方法,包括以下步骤:
步骤1、萨能奶山羊胎儿成纤维细胞的获得:
选取5只奶山羊做同期发情,与最后一次配种30天左右B超检查妊娠状况,选成功妊娠并与40-45天左右通过手术法无菌取出胎儿,用含双抗PBS冲洗三遍,放入无血清培养基快速带回实验室。在细胞室内无菌除去头、四肢、内脏,剩余皮肤组织酒精冲洗三遍,PBS冲洗三遍,然后剪成1mm3小块贴在细胞培养皿进行培养,每三天换一次培养基,待细胞铺满90%时消化传代,并作生长曲线及核型分析;
步骤2、RNA提取:
取对数生长期的细胞用trizol法提取RNA,该实验用到的所有物品包括1000mL、200μL、10μL tip头、枪头盒、Enpendoff管等均必须用0.1%的DEPC水或无RNA酶水浸泡6h以上或直接过夜,包装好后高压灭菌20-30min,65℃烘干备用;用到的所有研钵、玻璃器皿、金属器械等用锡箔纸包装好后放于180℃烘箱中烘烤8h以上,自然降温备用,提取的总RNA经微量分光光度计检查纯度及浓度,合适以后送华大基因进行高通量测序;
步骤3、文库构建及高通量测序
取上述总RNA经PAGE胶纯化、反转录,然后以HiSeq2000为测序平台进行高通量测序构建cDNA文库,建库过程及生物信息学分析流程如图1、图2;
具体操作过程依据Small RNA Sample Preparation Kit(Illumina,USA)试剂盒说明书步骤进行,简单过程如下:
(1)首先通过15%聚丙酰胺凝胶电泳分离总RNA,切割片段为18-30nt部分,然后进行分离纯化、胶回收;
(2)通过T4RNA连接酶分别在3’端和5’端添加3’((PO4)-CTGTAGGCACCATCAA-(NH2)-(CH2)6)和5’(ATCGTAGGCACCUGAAA)接头序列
(3)将成功添加接头序列的RNA进行反转录成cDNA,然后25个循环进行PCR扩增。
(4)90bp左右的PCR扩增产物经PAGE胶纯化回收以后其产物在深圳华大科技HiSeq2000平台进行测序。
(5)测序片段去除低质量序列、接头序列、重复序列以及污染序列后进行信息生物学分析。
步骤4、数据处理
萨能奶山羊胎儿成纤维细胞样品在HiSeq2000平台测序后得到冗余数据,对该原始数据首先要进行去低质量序列,去接头序列以及去污染序列等处理,获得干净序列即clean reads,然后再进行下一步生物信息学分析。其处理过程具体如下:
(1)去除有5’接头序列的reads;
(2)去除没有3’接头序列的reads;
(3)去除测序质量较低的reads;
(4)去除没有插入片段的reads;
(5)去除含有polyA的reads;
(6)去除小于18核苷酸的片段;
将获得的clean reads与绵羊全基因组数据库(International Sheep Genomics Consortium:ISGC)进行blast比对,用SOAPv1.11软件分析序列表达差异及分布情况,为了使每个unique sRNA有唯一的注释,我们采用如下优先规则:rRNAetc>known miRNA>repeat>exon>intron,由于rRNAetc是经Genbank数据库和Rfam数据库比对获得,实验规定两个数据库间的优先顺序为Genbank>Rfam,没有比对上任何注释信息的sRNA用unann表示。
为进一步分析候选miRNA二级结构,该实验用Mfold3.2软件]和RNAfold在线软件预测新miRNA颈环结构及最小自由能,用MIREAPv0.2软件再进一步深入分析每一个候选序列,参数设置如下:
(1)miRNA序列长度为18-26nt
(2)miRNA参考序列长度为20-24nt
(3)Drosha/Dicer酶切位点最少为3个
(4)miRNA在参考序列上最大拷贝数为20
(5)miRNA前体允许的最大自由能为200kcal/mol
(6)miRNA和miRNA*之间最大空格数为35
(7)miRNA和miRNA*之间最小配对数为14
(8)miRNA and miRNA*之间最大凸环为4。
以上所述,仅为本发明较佳的具体实施方式,本发明的保护范围不限于此,任何熟悉本技术领域的技术人员在本发明披露的技术范围内,可显而易见地得到的技术方案的简单变化或等效替换均落入本发明的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!