一种两性离子修饰的四苯乙烯及其制备方法和应用与流程
本发明涉及一种两性离子修饰的四苯乙烯及其合成方法和作为体异质结聚合物太阳能电池阴极界面修饰材料的用途。
背景技术:
近年来,体异质结(BHJ)聚合物太阳能电池(PSCs)发展迅速,随着在活性层材料开发、器件结构和界面工艺的优化方面取得的巨大进步,单节PSCs器件能量转换效率(PCE)已经突破11%。通常而言,PSCs器件结构是由活性层(共轭高分子给体与富勒烯衍生物受体)夹在阴极、阳极和相应的界面层之间组成的“三明治”结构,因此,如何通过改良界面修饰层减小电荷分离/收集的障碍以形成欧姆接触提升PCE近年来引起了科研人员越来越多的关注。
传统的无机金属化合物(氟化锂、碳酸铯、氟化铯等)阴极界面修饰材料(CILs)在器件制作过程中需要利用繁琐昂贵的真空蒸镀设备,相比之下,醇或水溶解性的有机小分子和聚合物CILs可以利用旋涂技术形成界面修饰层,并且CILs层与活性层迥异的溶解性可以避免后续旋涂过程对前一层形貌的影响,为潜在的卷对卷(roll-to-roll)式的大规模生产提供了可能。
目前已报道的醇或水溶解的CILs可以分为两类:π共轭和非π共轭的有机小分子或聚合物。π共轭的CILs主要由π共轭的主链和表面活性剂似的侧链构成,其中π共轭的单元主要是芴单元,侧链则有胺、季铵盐、磷酸盐、磺酸盐和两性离子盐等。已经报道的非π共轭的CILs则有两性离子盐、聚胺和氨基酸等。目前,研究人员对于界面修饰层背后的作用机理的认识依然不明确,两类共轭结构截然不同的CILs都有着不错的界面修饰效果;此外,由芴单元之外的π共轭单元构成的CILs材料的报道非常少,且固态下分子间会存在π-π堆积。因此,有必要开发一种新的构筑单元以拓展π共轭CILs的种类以及解决固态下分子间π-π堆积带来的不利影响。
技术实现要素:
为了解决现有技术中存在的技术难题,本发明通过将经典的聚集诱导发光分子四苯乙烯(TPE)与两性离子盐结合,提供了一种具备聚集诱导发光性质的醇或水溶解性的化合物,该化合物具有良好的界面修饰效果,可以用作体异质结聚合物太阳能电池阴极界面修饰材料。
本发明提供的技术方案具体如下:
一种两性离子修饰的四苯乙烯,具有式(I)或式(II)所示的结构:
一种上述两性离子修饰的四苯乙烯的合成方法,包括以下步骤:
(1)无水无氧条件下:将二苯甲烷溶于四氢呋喃中,0℃下将正丁基锂的己烷溶液滴入,0℃反应0.5小时后,向反应体系中加入4-甲氧基苯甲酰苯或二(4-甲氧基苯基)-甲酰,室温反应得到中间体粗产物,中间体粗产物与催化量的一水合对甲基苯磺酸一起在甲苯中回流,得白色固体状的化合物A,其结构式为
(2)无水无氧条件下:将化合物A溶于二氯甲烷中,降温至-78℃后滴入三溴化硼的二氯甲烷溶液,在-78℃反应1小时,然后自然升温至室温,继续反应,得白色固体状的化合物B,其结构式为
(3)无水无氧条件下:将化合物B和NaOH一起在丙酮中回流反应,然后加入N,N-二甲基-3-氯丙胺盐酸盐,继续回流,得白色固体状的化合物C,其结构式为
(4)室温下,化合物C和1,3-丙基磺酸内酯一起在乙酸乙酯中反应5天,反应完毕后,纯化,得白色固体状的两性离子修饰的四苯乙烯,其结构式为
步骤(1)中,二苯甲烷、正丁基锂和4-甲氧基苯甲酰苯或二(4-甲氧基苯基)-甲酰的投料摩尔比为5:5:4,反应在氮气保护下于Schlenk管中进行。
步骤(2)中,化合物A和三溴化硼的投料摩尔比为5:6,反应在氮气保护下于Schlenk管中进行。
步骤(3)中,化合物B、NaOH和N,N-二甲基-3-氯丙胺盐酸盐的投料摩尔比为2:6:3,反应在氮气保护下于Schlenk管中进行。
步骤(4)中,化合物C和1,3-丙基磺酸内酯的投料摩尔比为2:5。
上述两性离子修饰的四苯乙烯作为体异质结聚合物太阳能电池阴极界面修饰材料的应用。
本发明具有以下优点和有益效果:
(1)本发明第一次报道利用具有聚集诱导发光性质的化合物四苯乙烯构筑阴极界面修饰材料,既可以引入π单元提高其导电性,又可以避免固体分子间π-π堆积带来的不利影响。
(2)本发明在基于PTB7和PC71BM作为活性层、铝作为阴极的传统结构单层PSCs器件中表现出优秀的界面修饰效果,最高能量转换效率达到8.94%,比空白的铝电极器件PCE提高了2.3倍,比钙作为界面修饰层的铝电极器件PCE提高了21.6%。
附图说明
图1为化合物4的合成路线图。
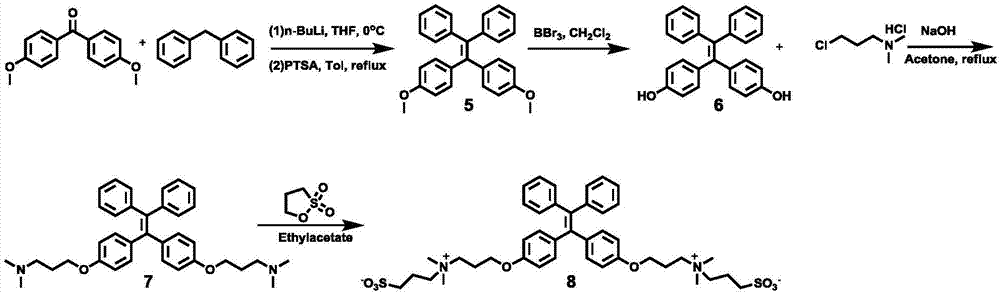
图2为化合物8的合成路线图。
图3为器件结构示意图。
具体实施方式
为了更好地理解本发明的内容,下面结合具体实施例对本发明的内容作进一步说明,但本发明的保护内容不局限于以下实施例。
本发明实施例中所用的原料可以由市场购得,或可用本领域已知的方法合成得到。
实施例1化合物1-8的合成
(1)氮气氛围下,在Schlenk管中加入二苯甲烷(3.36g,20mmol)和80mL干燥的四氢呋喃,在0℃滴入2.2M正丁基锂的己烷溶液(9.1mL,20mmol),于0℃反应0.5小时后向溶液中加入4-甲氧基苯甲酰苯(3.40g,16mmol),自然升温至室温后继续搅拌6小时。反应完毕后,加入饱和的氯化铵水溶液淬灭反应,用二氯甲烷萃取,收集有机相,用无水Na2SO4干燥,蒸出溶剂得到中间体粗产物。在250mL圆底烧瓶中将中间体溶解在80mL干燥的甲苯中,加入催化量的一水合对甲基苯磺酸(570mg,3.0mmol),回流12小时。反应完毕后冷却至室温,用10wt%的NaHCO3水溶液洗涤甲苯溶液,分液,收集有机相,用无水Na2SO4干燥,以石油醚为淋洗剂,将产品用硅胶色谱柱层析,分离纯化,真空干燥,得白色固体(5.93g,产率90.1%),并用1H NMR对结构进行表征,证实该白色固体为化合物1,其结构式为1H NMR(300MHz,CDCl3)δ(ppm):7.06(m,15H),6.94(d,J=8Hz,2H),6.64(d,J=8Hz,2H),3.74(s,3H).
(2)氮气氛围下,在Schlenk管中加入化合物1(1.08g,3.0mmol),加入10mL无水二氯甲烷将化合物1完全溶解,降温至-78℃后滴入三溴化硼的二氯甲烷溶液(1.7mL,3.6mmol),在-78℃反应1小时,然后自然升温至室温,继续反应12小时。反应完毕后,滴入适量冰水淬灭反应,有白色固体析出,抽滤,用去离子水洗滤饼3次,收集滤饼并真空干燥,得白色固体(1.02g,产率为98.1%),并用1H NMR,13C NMR和IR对结构进行表征,证实该白色固体为化合物2,其结构式为1H NMR(300MHz,CDCl3)δ(ppm):7.07(m,14H),6.90(d,J=8Hz,2H),6.57(d,J=8Hz,2H),4.60(s,1H).13C NMR(100MHz,THF-d8)δ(ppm):157.1,145.0,141.7,140.2,135.2,133.1,132.0,128.1,126.7,115.1.IR(KBr)υ(cm-1):3274(w),3054(w),3023(w),1605(w),1510(w),1492(w),1443(w),1265(w),1171(w),1102(w),1030(w),829(w).
(3)氮气氛围下,在Schlenk管中加入化合物2(696.8mg,2.0mmol)和NaOH固体(240mg,6mmol),加入15mL丙酮将化合物2完全溶解,回流0.5小时后,加入N,N-二甲基-3-氯丙胺盐酸盐(470mg,3,0mmol),继续回流36小时。反应完毕后,冷却至室温,抽滤并水洗滤渣,收集滤液,用二氯甲烷萃取,收集有机相,用无水Na2SO4干燥,以丙酮/三乙胺(v/v,10/1)为淋洗剂,将产品用硅胶色谱柱层析,分离纯化,真空干燥,得白色固体(480mg,产率55.1%),并用1H NMR,13C NMR和IR对结构进行表征,证实该白色固体为化合物3,其结构式为1H NMR(300MHz,CDCl3)δ(ppm):7.05(m,15H),6.91(d,J=8.7Hz,2H),6.51(d,J=8.7Hz,2H),3.93(t,J=6.3Hz,2H),2.42(t,J=7.2Hz,2H),2.24(s,6H),1.92(m,2H).13C NMR(75MHz,CDCl3)δ(ppm):157.7,144.2,140.7,140.2,136.2,132.7,131.5,127.8,126.5,113.8,66.1,56.6,45.6,27.6.IR(KBr)υ(cm-1):3054(w),3025(w),3054(w),2946(w),2862(w),2816(w),2766(w),1603(w),1506(w),1465(w),1443(w),1285(w),1244(w),1177(w),1055(w),1034(w),826(w).
(4)在50mL圆底烧瓶中加入化合物3(435.6mg,1mmol),加入3mL乙酸乙酯将化合物3完全溶解,将1,3-丙基磺酸内酯(305mg,2.5mmol)溶解在10mL的乙酸乙酯后滴入化合物3的溶液中,在室温下反应5天。反应完毕后,抽滤,收集滤饼用甲醇重结晶,得白色固体(443mg,79.8%),并用1H NMR,IR和HRMS对结构进行表征,证实该白色固体为化合物4,其结构式为1H NMR(300MHz,CD3OD)δ(ppm):7.04(m,15H),6.92(d,J=8.4Hz,2H),6.70(d,J=8.4Hz,2H),4.03(t,J=5.7Hz,2H),3.54(m,4H),3.13(s,6H),2.87(t,J=6.3Hz,2H),2.24(m,4H).IR(KBr)υ(cm-1):3415(w),3047(w),2926(w),2878(w),1602(w),1486(w),1442(w),1181(w),1042(w),824(w).HRMS(ESI,m/z):[M+Na]+calcd for C34H37O4NaS,578.2336;found,578.2341.
(5)化合物5的合成步骤和化合物1相同,仅将原料4-甲氧基苯甲酰苯换为二(4-甲氧基苯基)-甲酰,即得到白色粉末状的化合物5,产率为53.4%,其结构式为经过1H NMR表征,1H NMR(300MHz,CDCl3)δ(ppm):7.08(m,6H),7.02(m,4H),6.93(d,J=8.1Hz,4H),6.63(d,J=8.1Hz,4H),3.74(s,6H).
(6)化合物6的合成步骤和化合物2相同,化合物6:白色粉末,产率98.2%,其结构式为经过1H NMR表征,1H NMR(300MHz,DMSO-d6)δ(ppm):9.32(s,2H),7.10(m,6H),6.92(m,4H),6.73(d,J=8.1Hz,4H),6.48(d,J=8.1Hz,4H).
(7)化合物7的合成步骤和化合物3相同,化合物7:白色固体,产率63.1%,其结构式为经过1H NMR,13C NMR和IR表征,1H NMR(300MHz,CDCl3)δ(ppm):7.08(m,6H),7.02(m,4H),6.91(d,J=9.0Hz,4H),6.62(d,J=9.0Hz,4H),3.93(t,J=6.3Hz,4H),2.42(t,J=7.8Hz,4H),2.24(s,12H),1.90(m,4H).13C NMR(75MHz,CDCl3)δ(ppm):157.5,144.4,140.2,139.2,136.4,132.6,131.4,127.7,126.1,113.7,66.0,56.5,45.5,27.5.IR(KBr)υ(cm-1):3053(w),2946(w),2862(w),2816(w),2765(w),1604(w),1507(w),1465(w),1389(w),1285(w),1244(w),1176(w),1056(w),955(w),834(w).
(8)化合物8的合成步骤和化合物4相同,化合物8:白色固体,产率51.3%,其结构式为经过1H NMR,IR和HRMS表征,1H NMR(300MHz,CD3OD)δ(ppm):7.08(m,6H),6,98(m,4H),6.91(d,J=8.4Hz,4H),6.70(d,J=8.4Hz,4H),4.04(t,J=5.7Hz,4H),3.55(m,8H),3.13(s,12H),2.87(t,J=6.3Hz,4H),2.24(m,8H).IR(KBr)υ(cm-1):3423(w),3038(w),2928(w),2880(w),1603(w),1503(w),1288(w),1248(w),1205(w),1182(w),1062(w),1037(w),853(w).HRMS(ESI,m/z):[M+H]+calcd forC42H55O8S2,779.3395;found,779.3400.
本发明实施例中收集有机相后,用无水Na2SO4干燥,也可以采用其他干燥剂,只要能除去有机相中的水分且不与有机相反应即可。
实施例2:化合物4和8用作聚合物太阳能电池的阴极界面修饰材料的器件效果
以铝作为阴极,化合物4和8作为铝电极的界面修饰层,PTB7作为活性层给体材料,结构式为其中,R为PC71BM作为活性层受体材料,结构式为聚(3,4-亚乙二氧基噻吩)-聚(苯乙烯磺酸)(PEDOT:PSS)作为阳极的界面修饰层,结构式为ITO玻璃作为阳极。作为对比,也制作了不含阴极界面修饰层和钙作为阴极界面修饰层的器件。
本发明的化合物4和8用作体异质结聚合物太阳能电池的阴极界面修饰材料。以本发明的化合物4阴极界面修饰材料作为PTB7和PC71BM为活性层,铝作为阴极材料的传统结构单层PSCs器件最高能量转化效率达到8.27%,其对应的开路电压为0.76V,短路电流为16.02mA cm-2,填充因子为68.08%。以本发明的化合物8作为阴极界面修饰材料,PTB7和PC71BM为活性层,铝作为阴极材料的传统结构单层PSCs器件最高能量转化效率达到8.94%,比空白的铝电极器件(PCE=3.89%)提高了2.3倍,比钙作为界面修饰层的铝电极器件(PCE=7.35%)提高了21.6%,其对应的开路电压为0.76V,短路电流为16.86mAcm-2,填充因子为69.70%。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!