N,N’-双取代芳基硫脲衍生物及其合成方法和应用与流程
本发明属于药物合成技术领域,具体涉及一类N,N’-双取代芳基硫脲衍生物及其合成方法和应用。
背景技术:
硫脲类化合物具有广泛生物活性,如抗肿瘤、杀菌、抗结核、抗菌、抗甲状腺、抗病毒、抗癌抗肿瘤、抗HIV、抗炎、抗心律失常以及抗氧化及催眠和麻醉等多种活性。特别是其抗氧化和抗病毒等生物活性使硫脲类衍生物一度成为研究的热点。
首先,研究硫脲衍生物的抗氧化活性对后续抗肿瘤、抗炎等活性研究有着重要意义。硫脲类化合物的抗氧化活性使得该类化合物可用于阻止黑色素沉积,美白肌肤,如苯并咪唑氨基硫脲化合物是DPPH很好的清除剂(结构如下式)。异噁唑类硫脲化合物和噻唑硫脲类硫脲化合物都显示出不同程度的抗氧化活性。新型嘧硫脲衍生物具有抗氧化活性,尤其是氯苯,甲氧基取代的苯基硫脲衍生物具有很高抗氧化活性,硫脲衍生物的抗氧化活性明显强于脲衍生物(结构如下)。
另一方面,硫脲类化合物还具有广泛抗病毒活性,如嘌呤-6-甲醛缩氨基硫脲的抗疱疹病毒活性在上世纪六七十年代就已被人们揭示,随后揭示了双取代、单取代杂环都对单纯性疱疹病毒有抑制作用。杂环取代的硫脲类化合物还可作为HIV-1入侵时gp120与CD4结合的抑制剂(结构如下)。
硫脲类化合物还可作为HIV-1逆转录酶抑制剂(NNRTI),如已上市抗HIV药物的韦曲定(Trovirdine,LY300046·HCI)基本结构骨架就是硫脲;N-(2-苯乙基)-N'-(2-噻唑基)硫脲(PETT)类化合物及其衍生物PETT-1和PETT-2,PETT-3和PETT-4,PETT-5和PETT-6的结构中都含有苯乙基硫脲的结构。其中,PETT-3和PETT-4对III B和ROD的抑制活性和选择性比较强,其EC50和CC50浓度在10μg/mL,并具有很强抗菌活性。PETT-5和PETT-6能够减弱HIV-1引起MT-4细胞的病变和抑制MT-4细胞生长,并且具有选择性。从化合物库中筛选得到的化合物PETT-7及其后续结构改造衍生物PETT-8对HIV-1RT具有抑制作用,后者在5mmol/L浓度下对HIV-1假病毒的抗感染活性IC50为1.1μM(结构如下)。
与PETT类化合物类似,亚氨基取代的硫脲衍生物(ITU)对HIV-1RT也表现出不同程度的抑制活性,如化合物ITU-2和ITU-3的抑制活性pIC50分别为-1.111和-1.360(结构如下)。
磺酰胺双苯基硫脲衍生物(SDBTU)HIV-1对病毒衣壳蛋白的合成具有抑制作用,进而可影响HIV-1复制。其中SDBTU-1和SDBTU-2具有较高抑制活性,IC50分别为0.35μM和0.50μM。进一步结构改造得苯基嘧啶双取代硫脲衍生物BPTU-1和BPTU-2,不仅具有抑制HIV衣壳蛋白的活性,还具有抗人类亲环素A的活性(结构如下)。
在前期研究证明了含有吲唑取代的不对称芳基硫脲类具有一定抗病毒和抗氧化活性,因此,本发明在上述研究基础上对其进行结构简化,进一步设计合成一系列新型结构的不对称芳基硫脲类目标化合物,并测试所有目标化合物的抗氧化活性和抗病毒活性。
技术实现要素:
本发明的目的在于提供一类N,N’-双取代芳基硫脲衍生物及其合成方法和应用。
本发明是通过以下技术方案来实现:
本发明公开了一类N,N’-双取代芳基硫脲衍生物,其结构式如下:
其中,R1为H、甲基;R2为H、F、Cl或CF3;R3为H、F、Cl、Br、甲基、甲氧基、乙炔基或NHSO2Me;R4为H、F、Cl、Br、甲基、甲氧基或NHSO2Me;R5为H;R6为H,X为C或N。
本发明还公开了上述N,N’-双取代芳基硫脲衍生物的制备方法,包括以下步骤:
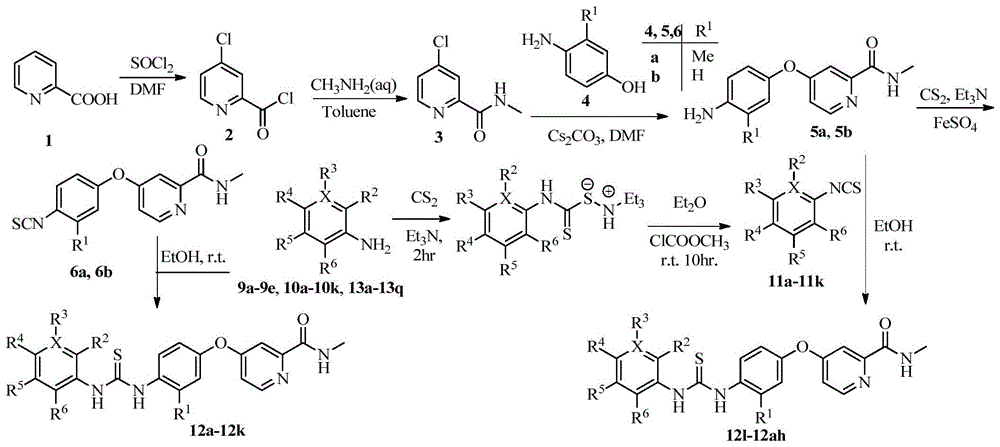
1)以2-吡啶甲酸(1)为原料,于72℃下,在氯化亚砜中反应生成4-氯吡啶-2-甲酰氯(2);
2)4-氯吡啶-2-甲酰氯(2)与甲胺水溶液在20℃以下进行反应,再经碱化生成N-甲基-4-氯吡啶-2-甲酰胺(3);
3)在碳酸铯的存在下,N-甲基-4-氯吡啶-2-甲酰胺(3)与4-氨基苯酚(4b)或3-甲基-4-氨基苯酚(4a),于105℃下,发生威廉姆森成醚反应,生成4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5b)或4-(3-甲基-4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5a);
4)将4-(3-甲基-4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5a)或4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5b)在二硫化碳、三乙胺作用下生成其三乙胺盐,然后再在硫酸亚铁的转化下,反应生成N-甲基-4-(3-甲基-4-异硫氰酸酯苯氧基)吡啶-2-甲酰胺(6a)或N-甲基-4-(4-异硫氰酸酯苯氧基)吡啶-2-甲酰胺(6b);
5)以二氯甲烷作溶剂,将硝基取代苯胺(7)在弱碱作用下与甲磺酰氯反应,生成苯甲磺酰胺(8),然后将苯甲磺酰胺(8)溶于乙醇,且在氯化亚锡的还原作用下,将硝基还原成氨基,可制得不同取代的氨基苯甲磺酰胺(9);
6)将苯胺10a或苯胺衍生物在二硫化碳、三乙胺作用下,生成三乙胺盐中间体,然后在氯甲酸甲酯转化下,生成取代苯异硫氰酸酯(11);
7)目标化合物12是由相应的异硫氰酸酯和取代芳香胺中乙醇中室温反应制备,制备方法由下列方法之一:
方法一:将N-甲基-4-(3-R1-4-异硫氰酸酯苯氧基)吡啶-2-甲酰胺(6a,R1=CH3或6b,R1=H)和取代芳胺(13a~13q)溶解于无水乙醇中,室温搅拌反应,制备目标化合物12a~12k;
方法二:将各种取代芳异硫氰酸酯(11a~11k)和4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺及其甲基衍生物(5a,5b)溶解于无水乙醇中,室温搅拌反应,制备目标化合物12l~12h。
步骤1)中,2-吡啶甲酸(1)与氯化亚砜的用量比为1g:3~5mL;甲胺与2-吡啶甲酸(1)的质量比为3~4:1;步骤3)中,N-甲基-4-氯吡啶-2-甲酰胺(3)、4-氨基苯酚(4b)或3-甲基-4-氨基苯酚(4a)及碳酸铯的反应摩尔比为:(7~10):(8~10):(14~15)。
步骤4)中,4-(3-甲基-4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5a)或4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5b)、二硫化碳及三乙胺的用量比为(400~620)mg:(160~230)mg:2mL。
步骤5)中,硝基取代苯胺7为间硝基苯胺、对硝基苯胺、2-甲基-5-硝基苯胺、2-甲基-4-硝基苯胺或2-甲氧基-4-硝基苯胺;且硝基取代苯胺7与甲磺酰氯的摩尔比为(1~12):91.6。
步骤6)中,苯胺衍生物为邻氟苯胺、对氟苯胺、邻氯苯胺、对氯苯胺、间溴苯胺、对溴苯胺、2,3-二氯苯胺、对甲氧基苯胺或对乙炔基苯胺;且苯胺或苯胺衍生物、二硫化碳、三乙胺及氯甲酸甲酯的反应用量比为(2.3~6.5)g:3.6mL:7.2mL:3.9mL。
本发明还公开了上述N,N’-双取代芳基硫脲衍生物在制备抗氧化药物中的应用。
本发明还公开了上述N,N’-双取代芳基硫脲衍生物在制备抗病毒药物中的应用。
所述抗病毒药物包括抗疱疹病毒、抗牛痘病毒、抗呼吸肠道病毒、抗Coxsackie病毒、抗Feline冠状疱疹病毒及抗HIV病毒的药物。
与现有技术相比,本发明具有以下有益的技术效果:
本发明公开了一类N,N’-双取代芳基硫脲衍生物,是一系列同时含有各种取代芳环结构和不对称取代硫脲结构的化合物,均为未见文献报道的新型结构化合物。结合目标化合物骨架结构及官能团的特点,本发明根据重要中间体异硫氰酸酯的性质,决定了总的合成路线将从两个方面完成:二硫化碳与芳胺在叔胺(如三乙胺、吡啶)作用下反应先生成芳胺二硫代氨基甲酸盐,然后在氯甲酸甲酯作用下生成异硫氰酸酯,是制备不同取代的芳基异硫氰酸酯最为普遍的方法。但是,当苯胺其他位置上连接有强吸电子基(如硝基、氰基、三氟甲基等)或者多个取代基时,采用该反应产率低、选择性差,甚至不能发生反应。此时,只能将中间体5,在三乙胺作用下,换用硫酸亚铁作为转化试剂生成6,化合物6再与苯胺缩合反应生成目标化合物12。
本发明通过对所有目标化合物的生物活性测试分析包括DPPH抗氧化活性、抗病毒活性测定。结果表明,多数目标化合物都具有较好的抗氧化活性,表现出对DPPH具有较高清除率。所有目标化合物的DPPH清除率几乎都超过了50%,表现出不同的抗氧化活性。清除率较高的化合物有12q,12ae和12r清除率都达到90%。其中化合物12q抗氧化活性活性较好,对DPPH的清除活性IC50为0.025mg/mL,活性与阳性对照的抗坏血酸相近,甲磺酰胺基产物活性也较高,12ae的IC50为0.12mg/mL。化合物12h,12j,12y和12ag具有一定的抗疱疹病毒活性,可抑制单纯疱疹病毒-1(KOS)、单纯疱疹病毒-2(G)活性,且细胞毒性较弱。分析上述化合物的构效关系可知:吡啶取代的硫脲衍生物的抑制活性要比苯基取代的要高。A环为硝基苯取代的硫脲衍生物活性较高,但比吡啶取代物活性要差。总之,本专利所设计合成的化合物结构新颖,其抗氧化活性和抗病毒活性首次被揭示。另外有望还有未知的生物活性未充分阐明,可为进一步新药研发提供一定物质基础。
附图说明
图1为本发明的目标化合物的合成路线图;
图2为本发明的中间体氨基苯甲磺酰胺的合成路线图;
图3为化合物12j,12r对DPPH清除率曲线图;其中,(a)为12j浓度与清除率曲线;(b)为12r对DPPH清除率曲线。
具体实施方式
下面结合具体的实施例对本发明做进一步的详细说明,所述是对本发明的解释而不是限定。
一、合成部分
本发明的目标化合物的合成路线图,参见图1:
1、N-甲基-4-(3-R1-4-异硫氰酸酯苯氧基)吡啶-2-甲酰胺
1)N-甲基-4-氯吡啶-2-甲酰胺(3)
量取氯化亚砜(40.0mL)于250mL干净烧瓶中,向其中滴加干燥的DMF(2.0mL),并加热至50℃,搅拌20min。然后向烧瓶中分批加入2-吡啶甲酸(1,10.0g,81.4mmol)(分5次,每次2.00g,30min内加完),体系由绿色变为黄绿色最后变为亮红色。然后升温至72℃,回流下搅拌反应18hrs,体系有灰黄色固体生成。加入甲苯(50.0mL),减压蒸馏至10.0mL,该步骤重复2次,除去过量氯化亚砜。加入甲苯(10.0mL),得到4-氯吡啶-2-甲酰氯(2)的甲苯溶液,直接用于下步反应。
取甲胺水溶液(30%,32.0g),冰乙醇浴下冷却至-5℃,控制温度在20℃以下滴加上述的甲苯溶液,滴毕,同温反应1.5hrs,转移至分液漏斗中静置分层,甲苯层用饱和食盐水(20.0mL×2)洗涤,得到甲苯层,减压下旋蒸除有机溶剂,剩余棕色油状物加入THF(30.0mL),冷却至5℃,20℃以下加入36%的盐酸(10.0g),加毕同温度下反应1hr,有大量红褐色固体。过滤体系,得暗红色滤饼,用THF(10.0mL×2)洗涤,然后滤饼溶于水(20.0mL),冰浴条件加入20%NaOH溶液调pH至7-8,有大量固体产生,搅拌30mins,过滤,滤饼用水洗涤(10.0mL×2),于20℃下真空干燥得黄色固体(7.82g,56.6%),两步m.p.35-36℃(lit.m.p.38-39℃)。2)4-(3-甲基-4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5a)
将化合物4a(2.00g,16.26mmol)溶于无水DMF(20.0mL)中,加入碳酸铯(9.26g,28.5mmol),于50℃和氮气保护下搅拌1hr,体系颜色逐渐由黄变绿。然后滴加化合物3(2.50g,14.7mmol)的DMF(10.0mL)溶液,加热到105℃,氮气保护下搅拌反应8hr,TLC监测至原料完全反应。待溶液冷却后,过滤出碳酸铯固体,加入乙酸乙酯(100mL)洗涤滤饼得棕褐色滤液。滤液中加入饱和氯化钠溶液(100mL),水层被分出,用乙酸乙酯(60.0mL×3)萃取,合并有机相,用饱和氯化钠水溶液(4×100mL)洗涤,得到的有机相再用无水硫酸钠干燥,过滤除干燥剂,减压下回收溶剂,得棕色油状物。柱层析分离(石油醚:乙酸乙酯v/v=2:1~1:2体系梯度洗脱),干燥得灰棕色固体(2.83g,73.4%),m.p.128.5-129.3℃,MS:258.0。1H NMR(400MHz,DMSO-d6)δ:8.60(s,1H,pyridinyl-H),8.45(d,J=5.6Hz,1H,pyridinyl-H),7.33(d,J=2.6Hz,1H,pyridinyl-H),7.07(q,J=5.6,2.6Hz,1H,CH3-NH),6.75(ddd,J=26.2,19.3,5.5Hz,1H,Ar-H),4.94(s,2H,Ar-NH2),2.78(d,J=4.9Hz,3H,N-CH3),2.08(s,3H,Ar-CH3)。
3)4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5b)
将化合物4b(2.40g,20.0mmol)溶于无水DMF(30.0mL)中,加入碳酸铯(9.75g,30.0mmol),于50℃下,氮气保护下,搅拌1hr,体系由黄色变为黄绿色。然后向体系滴加化合物3(3.40g,20.0mmol)的DMF(10.0mL)溶液,加热至105℃,氮气保护下搅拌反应9hr。TLC监测反应至完全,过滤出固体,减压下旋蒸出大部分的DMF,加入乙酸乙酯(200mL),饱和氯化钠溶液(200mL),水层被分出,用乙酸乙酯(100mL)萃取两次,合并有机相,用饱和氯化钠溶液(100mL)洗涤有机层3次,合并有机相,无水硫酸钠干燥。旋蒸出有机溶剂,将得到的油状物溶于THF(30.0mL),于低于20℃的条件下,缓慢滴加滴加浓盐酸(9.00g),有大量固体产生,搅拌1hr,过滤得浅灰棕色固体,用THF(10.0mL)洗涤两次,将得到的固体溶于水(30.0mL)中,滴加20%的NaOH溶液调pH至7-8,有大量固体析出,大力搅拌30mins,过滤得浅灰棕色固体,水洗两遍,固体减压下干燥得浅灰色固体(2.24g,46.2%),m.p.99-101℃。MS:243.1,186.1,109.1。
4)4-(3-甲基-4-异硫氰酸酯苯氧基)-N-甲基-2-吡啶甲酰胺(6a)
向100mL圆底烧瓶中依次加入化合物5a(409mg,1.60mmol),二硫化碳(160mg,2.00mmol),三乙胺(2.0mL),丙酮(10.0mL)。氮气保护下,室温下搅拌反应1.5hr,体系呈红色,再向体系中加入少量的三乙胺、硫酸亚铁,体系的颜色瞬间变为暗红色,继续搅拌反应6h,TLC检测原料反应完全。柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱),旋蒸干燥后得类白色固体(310mg,64.9%),m.p.128.0-129.1℃。1H NMR(400MHz,DMSO-d6)δ:8.81(s,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.53(d,J=8.6Hz,1H,Ar-H),7.41(d,J=2.5Hz,1H,pyridinyl-H),7.28(d,J=2.6Hz,1H,Ar-H),7.20(q,J=5.6,2.6Hz,1H,CH3-NH),7.15(dd,J=8.6,2.6Hz,1H,Ar-H),2.79(d,J=4.9Hz,3H,N-CH3),2.38(s,3H,Ar-CH3)。
5)4-(4-异硫氰酸酯苯氧基)-N-甲基-2-吡啶甲酰胺(6b)
向100mL圆底烧瓶中依次加入化合物5b(608mg,2.50mmol),二硫化碳(228mg,3.00mmol),三乙胺(2.0mL),丙酮(5.0mL)。室温下搅拌反应1hr,此时体系颜色呈鲜红色。再向体系中加入少量的三乙胺、硫酸亚铁,体系的颜色瞬间变为暗红色,继续搅拌反应6h,减压蒸馏,除去过量的二硫化碳及溶剂,向剩余物中加入冰水淬灭,搅拌一段时间使体系分散均匀后,再向其中加入大量乙酸乙酯充分溶解,过滤出黑色固体,得滤液。用饱和食盐水(100mL)洗涤三次有机层,合并有机相。柱层析分离(洗脱剂石油醚/乙酸乙酯v/v=2:1-1:1梯度洗脱),干燥后得得类白色固体(468mg,65.7%),m.p.124.0-126.0℃。MS:285.0,228.1。1H NMR(400MHz,DMSO-d6)δ:2.79(d,3H,N-CH3),7.20(q,1H,CH3-NH),7.33(d,2H,Ar-H),7.42(s,1H,pyridinyl-H),7.59(d,2H,Ar-H),8.54(d,1H,pyridinyl-H),8.81(d,1H,pyridinyl-H)。
2、不同取代氨基苯甲磺酰胺化合物的制备
中间产物氨基苯甲磺酰胺的合成路线图参见图2:
1)硝基苯甲磺酰胺的制备
(1)间硝基苯甲磺酰胺(8a)的制备
取化合物间硝基苯胺7a(1.00g,6.58mmol),溶解于干燥的二氯甲烷(15.0mL)中,冰浴条件下缓慢滴加甲磺酰氯(7.0mL,91.6mmol),将吡啶(3.0mL)的二氯甲烷溶液滴加至上述体系中,氮气保护下,控制体系温度不高于20℃,搅拌反应2hrs后恢复室温过夜反应,体系中有少量的白色固体析出,TLC监控原料几乎完全反应。过滤出体系中的固体,用二氯甲烷(50.0mL)洗涤固体,得到有机层。向有机层中加入水(100mL×3),转移至分液漏斗中,萃取得下层的有机层,合并有机相,减压下旋蒸出大部分二氯甲烷,乙酸乙酯/石油醚重结晶过滤得白色晶体8a(0.620g,46.5%),m.p.162.3-163.4℃(Lit.m.p.164-165℃)。
(2)4-硝基苯甲磺酰胺(8b)的制备
称取化合物对硝基苯胺7b(230mg,1.00mmol)溶于二氯甲烷(25.0mL)中,冰浴条件下缓慢滴加甲磺酰氯(7.0mL,91.6mmol),将吡啶(3.0mL)的二氯甲烷溶液滴加至上述体系中,操作同化合物8a的合成,乙醇重结晶得白色粉末8b(620mg,46.0%),m.p.180.0-181.3℃(Lit.m.p.182.5-183.5℃)。
(3)2-甲基-5-硝基苯甲磺酰胺(8c)的制备
称取化合物2-甲基-5-硝基苯胺7c(1.00g,6.58mmol)溶于二氯甲烷(15.0mL)中,冰浴条件下缓慢滴加甲磺酰氯(7.0mL,91.6mmol),将吡啶(6.0mL)的二氯甲烷溶液滴加至上述体系中,操作同化合物8a的合成,TLC监控不再发生反应,乙酸乙酯/石油醚重结晶过滤得白色粉末8c(1.00g,64.0%),m.p.153.9-155.1℃(Lit.m.p.155-156℃)。
(4)2-甲基-4-硝基苯甲磺酰胺(8d)的制备
称取化合物2-甲基-4-硝基苯胺7d(1.00g,6.58mmol)溶于二氯甲烷(35.0mL)中,冰浴条件下缓慢滴加甲磺酰氯(7.0mL,91.6mmol),将吡啶(6.0mL)的二氯甲烷溶液滴加至上述体系中,操作同化合物8a的合成,向旋蒸得到的黄色油状物中加入乙酸乙酯/石油醚重结晶过滤得白色粉末8d(800mg,57.6%),m.p.193.7-194.8℃。
(5)2-甲氧基-4-硝基苯甲磺酰胺(8e)的制备
称取化合物2-甲氧基-4-硝基苯胺7e(2.00g,11.9mmol)溶于二氯甲烷(25.0mL)中,冰浴条件下缓慢滴加甲磺酰氯(7.0mL,91.6mmol),将吡啶(6.0mL)的二氯甲烷溶液滴加至上述体系中,操作同化合物8a的合成,旋蒸得到的棕褐色固体,乙酸乙酯/石油醚重结晶过滤得金黄色粉末8e(1.96g,68.0%),m.p.108.5-109.0℃(Lit.m.p.109-110℃)。
2)氨基苯甲磺酰胺的制备
(1)3-氨基苯甲磺酰胺(9a)的制备
取8a(558mg,3mmol)溶解于无水乙醇(30.0mL)中,加入氯化亚锡(5.0equiv.),于70℃下,搅拌反应6h,TLC监测至反应完全,冷却至室温,用5%的碳酸氢钠溶液调pH至9-10,体系中出现大量黄色絮状物,用乙酸乙酯萃取,有机相用饱和食盐水洗涤,分出有机相,用无水硫酸钠干燥,过滤得滤液,蒸出有机试剂,得黄色油状物,向其中加入乙酸乙酯、石油醚调节体系极性,出现大量固体,过滤得黄色粉末9a(210mg,41.7%),m.p.127.4-128.8℃(Lit.129-130℃)。
(2)4-氨基苯甲磺酰胺(9b)的制备
取8b(558mg,2.60mmol)溶解于无水乙醇(25.0mL)中,加入氯化亚锡(5.0equiv.),于70℃下,搅拌反应6h,操作同8a的合成,旋蒸出溶剂出现大量固体,乙酸乙酯/石油醚重结晶过滤得黄色粉末9b(0.33g,75.6%),m.p.116.3-117.6℃(Lit.114-115℃)。
(3)2-甲基-5-氨基苯甲磺酰胺(9c)的制备
取8c(690mg,3.00mmol)溶解于无水乙醇(35.0mL)中,加入氯化亚锡(5.0equiv.),于70℃下,搅拌反应6h,操作同8a的合成,旋蒸出溶剂出现大量固体,乙酸乙酯/石油醚重结晶过滤得黄色粉末9c(0.57g,95.1%),m.p.135.0-136.7℃(Lit.137.5-138.5℃)。
(4)2-甲基-4-氨基苯甲磺酰胺(9d)的制备
取8d(690mg,3.00mmol)溶解于无水乙醇(40.0mL)中,加入氯化亚锡(5.0equiv.),于70℃下,搅拌反应6h,操作同8a的合成,旋蒸出溶剂出现大量固体,向其中加入石油醚有大量絮状物析出,过滤得白色粉末9d(0.350g,58.7%),m.p.142.1-143.3℃(Lit.142-143℃)。
(5)2-甲氧基-4-氨基苯甲磺酰胺(9e)的制备
取8e(490mg,2.00mmol)溶解于无水乙醇(20.0mL)中,加入氯化亚锡(5.0equiv.),于70℃下,搅拌反应6h,操作同8a的合成,旋蒸出溶剂出现大量固体,向其中加入石油醚发现有大量固体析出,过滤得暗红色粉末9e(0.250g,58.7%),m.p.186.3-187.6℃。
3、不同取代的苯异硫氰酸酯的制备
1)苯异硫氰酸酯(11a)的制备
称取苯胺10a(4.65g,50.0mmol)置于100mL圆底烧瓶,依次加入二硫化碳(3.6mL,90.0mmol)、乙醚(10.0mL)、以及三乙胺(7.2mL),反应体系于35℃下搅拌12h。体系产生大量固体,抽滤,无水乙醚(30.0mL)洗涤滤饼,得浅黄色粉末状固体。将该固体置空气中自然干燥30min,挥去其中残存的乙醚后,将其转移至100mL干净圆底烧瓶中,加氯仿(50.0mL),体系成均相,加入三乙胺(7.2mL),冰盐浴冷却至0℃以下,搅拌下向体系中滴加氯甲酸甲酯(3.9mL,50.0mmol),滴加过程中,体系温度控制在5℃以下。滴加完毕,水浴29~30℃反应1hr,停止反应,柱层析分离(石油醚体系),得浅黄色油状液体11a(6.10g,90.2%)。
2)邻氟苯异硫氰酸酯(11b)的制备
称取2-氟苯胺10b(5.55g,50.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11b(3.8g,50.3%)。
3)对氟苯异硫氰酸酯(11c)的制备
称取4-氟苯胺10c(5.55g,50.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11c(3.1g,41.0%)。
4)邻氯苯异硫氰酸酯(11d)的制备
称取2-氯苯胺10d(6.35g,50.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11d(2.5g,62.4%)。
5)对氯苯异硫氰酸酯(11e)的制备
称取4-氯苯胺10e(6.35g,50.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得白色固体11e(7.9g,94.1%),m.p.41.8-42.9℃(Lit.m.p.46-47℃)。
6)间溴苯异硫氰酸酯(11f)的制备
称取3-溴苯胺10f(3.40g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11f(3.25g,76.3%)。
7)对溴苯异硫氰酸酯(11g)的制备
称取4-溴苯胺10g(3.40g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得白色固体11g(3.40g,80.5%),m.p.51.2-52.2℃(lit.m.p.53.5-54.5℃)。
8)2,3-二氯苯异硫氰酸酯(11h)的制备
称2,3-二氯苯胺10h(2.80g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11h(1.58g,43.2%)。
9)对甲氧基苯异硫氰酸酯(11i)的制备
称4-甲氧基苯胺10i(2.50g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得白色固体11i(3.0g,90.4%),m.p.21.1-22.2℃。10)对乙炔基苯异硫氰酸酯(11j)的制备
称4-乙炔基苯胺10j(2.30g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12hrs。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11j(1.30g,41.1%)。
11)邻三氟甲基苯异硫氰酸酯(11k)的制备
称4-乙炔基苯胺10k(3.20g,20.0mmol)置于一干净100mL圆底烧瓶,依次加入乙醚(10.0mL)、二硫化碳(3.6mL,90.0mmol)以及三乙胺(7.2mL),体系于35℃下反应12h。操作同11a的合成,柱层析分离(石油醚体系),得浅黄色油状液体11k(0.35g,8.7%)。
4、N-取代苯基-N’-[2-R1-4-[2-(N-甲基胺基甲酰基)-4-吡啶基氧基]苯基]硫脲(12)的制备
将化合物11(a~k)和化合物5a(1equiv.)溶解于乙醇中,氮气保护下室温反应,搅拌过夜,TLC监测至反应完全,减压旋蒸出大部分乙醇,向体系中加入适量冰水,搅拌半小时后,过滤得固体初产物,重结晶或者通过柱层析分离得目标化合物,利用此法制备12a-12k。
1)N-苯基-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12a)的制备
取化合物11a(135mg,1.00mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将5a(257mg,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,氮气保护下室温反应过夜,TLC监控完全反应,减压旋蒸出大部分乙醇,向体系中加入适量冰水,搅拌0.5hr后,过滤得固体初产物,石油醚/乙酸乙酯重结晶(v/v=4:1)得灰色固体12a(150mg,38.3%),m.p.171.4-172.4℃。1H NMR(400MHz,DMSO-d6)δ:9.78(s,1H,thiourea-H),9.37(s,1H,thiourea-H),8.79(s,1H,pyridinyl-H),8.54(d,J=5.2Hz,1H,pyridinyl-H),7.49(d,J=5.7Hz,2H,Ar-H),7.44(s,1H,pyridinyl-H),7.37(m,3H,Ar-H),7.20–7.14(m,3H,Ar-H),7.05(s,1H,CH3-NH),2.80(d,J=2.2Hz,3H,NH-CH3),2.28(s,3H,Ar-CH3)。
2)N-(2-氟苯基)-N'-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12b)的制备
取化合物11b(155mg,1.00mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将5a(257mg,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶得灰红色固体12b(267mg,65.1%),m.p.168.4-168.8℃。1H NMR(400MHz,DMSO-d6)δ:9.55(s,1H,thiourea-H),9.43(s,1H,thiourea-H),8.80(d,J=4.8Hz,1H,pyridinyl-H),8.53(d,J=5.6Hz,1H,pyridinyl-H),7.57(t,J=7.8Hz,1H,Ar-H),7.45(d,J=2.5Hz,1H,pyridinyl-H),7.37(d,J=8.5Hz,1H,Ar-H),7.27(dd,J=9.3,2.6Hz,2H,Ar-H),7.18(m,J=10.2,8.0,3.0Hz,3H,Ar-H),7.06(q,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.28(s,3H,Ar-CH3)。
3)N-(4-氟苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12c)的制备
取化合物11c(200mg,1.30mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将5a(257mg,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶得灰色固体12c(150mg,28.1%),m.p.141.4-142.3℃。1H NMR(400MHz,DMSO-d6)δ:9.70(s,1H,thiourea-H),9.37(s,1H,thiourea-H),8.79(d,J=4.7Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.49–7.43(m,3H,Ar-H),7.37(d,J=8.5Hz,1H,pyridinyl-H),7.21–7.13(m,4H,Ar-H),7.06(q,J=8.5,2.6Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
4)N-(2-氯苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12d)的制备
取化合物11d(200mg,1.18mmol)溶解于无水乙醇(20.0mL)置于100mL圆底烧瓶中,将5a(0.257g,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶得红色固体12d(300mg,70.4%),m.p.171.9-172.3℃。1H NMR(400MHz,DMSO-d6)δ:9.53(s,1H,thiourea-H),9.39(s,1H,thiourea-H),8.82–8.78(m,1H,pyridinyl-H),8.53(d,J=5.6Hz,1H,pyridinyl-H),7.58(d,J=6.7Hz,1H,pyridinyl-H),7.52(dd,J=7.9,1.4Hz,1H,Ar-H),7.39(m,3H,Ar-H),7.29(dd,J=7.7,1.6Hz,1H,Ar-H),7.21–7.15(m,2H,Ar-H),7.07(dd,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.9Hz,3H,CONH-CH3),2.29(s,3H,Ar-CH3)。
5)N-(4-氯苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12e)的制备
取化合物11e(100mg,0.59mmol)溶解于无水乙醇(20.0mL)置于100mL圆底烧瓶中,将5a(150mg,0.60mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶得白色固体12e(160mg,59.0%),m.p.159.8-160.1℃。1H NMR(400MHz,DMSO-d6)δ:9.84(s,1H,thiourea-H),9.47(s,1H,thiourea-H),8.81(d,J=4.8Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.53(d,J=8.8Hz,1H,pyridinyl-H),7.44(d,J=2.5Hz,1H,Ar-H),7.38(dd,J=10.3,8.8Hz,3H,Ar-H),7.19(dd,J=5.6,2.6Hz,3H,Ar-H),7.16(d,J=2.6Hz,1H,Ar-H),7.06(dd,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
6)N-(3-溴苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12f)的制备
取化合物11f(250mg,1.00mmol)溶解于无水乙醇(15.0mL)置于100mL圆底烧瓶中,将5a(257mg,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶得白色固体12f(0.36g,77.0%),m.p.143.1-143.3℃。1H NMR(400MHz,DMSO-d6)δ:9.88(s,1H,thiourea-H),9.56(s,1H,thiourea-H),8.81(d,J=4.9Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.82(s,1H,pyridinyl-H),7.48–7.42(m,2H,Ar-H),7.37(d,J=8.5Hz,1H,Ar-H),7.30(dd,J=12.5,4.9Hz,2H,Ar-H),7.22–7.14(m,2H,Ar-H),7.07(dd,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.9Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
7)N-(4-溴苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12g)的制备
取化合物11g(210mg,1.00mmol)溶解于无水乙醇(15.0mL)置于100mL圆底烧瓶中,将5a(257mg,1.00mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶得褐色固体12g(360mg,36.0%),m.p.162.5-163.4℃。1H NMR(400MHz,DMSO-d6)δ:9.84(s,1H,thiourea-H),9.48(s,1H,thiourea-H),8.80(d,J=4.9Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.54–7.46(m,4H,Ar-H),7.44(d,J=2.5Hz,1H,pyridinyl-H),7.37(d,J=8.6Hz,1H,Ar-H),7.19(dd,J=5.6,2.6Hz,1H,Ar-H),7.16(d,J=2.6Hz,1H,Ar-H),7.06(dd,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.9Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
8)N-(2-甲基-3-二氯苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12h)的制备
取化合物11h(132mg,0.72mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将5a(0.19g,0.72mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=3:1)重结晶得灰褐色固体12h(280mg,88.2%),m.p.96.6-97.4℃。1H NMR(400MHz,CDCl3)δ:8.43(d,J=5.5Hz,1H,thiourea-H),8.06(s,1H,thiourea-H),7.69(d,J=2.4Hz,2H,pyridinyl-H),7.45(d,J=8.5Hz,1H,Ar-H),7.41(d,J=7.9Hz,1H,pyridinyl-H),7.32(d,J=7.7Hz,1H,CONHCH3),7.29–7.20(m,3H,Ar-H),7.06–6.99(m,3H,Ar-H),3.01(d,J=5.1Hz,3H,CONH-CH3),2.40(s,3H,ClPh-CH3),2.34(s,3H,Ar-CH3)。
9)N-(4-甲氧基苯基)-N'-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12i)的制备
取化合物11i(400mg,2.40mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将5a(190mg,0.72mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶得灰褐色固体12i(280mg,42.0%),m.p.187.7-188.1℃。1H NMR(400MHz,DMSO-d6)δ:9.56(s,1H,thiourea-H),9.21(s,1H,thiourea-H),8.80(d,J=4.9Hz,1H,pyridinyl-H),8.53(d,J=5.6Hz,1H,pyridinyl-H),7.44(d,J=2.5Hz,1H,pyridinyl-H),7.36(d,J=8.6Hz,1H,Ar-H),7.34–7.30(m,2H,Ar-H),7.18(dd,J=5.6,2.6Hz,1H,Ar-H),7.14(d,J=2.6Hz,OH),7.04(dd,J=8.5,2.7Hz,1H,CONHCH3),6.92(d,J=8.9Hz,1H),3.75(s,3H,OCH3),2.79(d,J=4.8Hz,1H,CONH-CH3),2.26(s,3H,Ar-CH3)。
10)N-(3-乙炔苯基)-N'-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12j)的制备
取化合物11j(100mg,0.60mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将5a(160mg,0.6mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶得灰褐色固体12j(220mg,88.1%),m.p.87.3-88.6℃。1H NMR(400MHz,DMSO-d6)δ:9.82(s,1H,thiourea-H),9.51(s,1H,thiourea-H),8.80(d,J=4.7Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.65(s,1H,pyridinyl-H),7.51(d,J=8.2Hz,1H,Ar-H),7.44(d,J=2.5Hz,1H,Ar-H),7.40–7.31(m,2H,Ar-H),7.24(d,J=7.6Hz,1H,Ar-H),7.20–7.13(m,2H,Ar-H),7.06(dd,J=8.6,2.7Hz,1H,CONHCH3),4.21(s,1H,ethyne-H),2.79(d,J=4.8Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
11)N-(2-三氟甲基苯基)-N'-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12k)的制备
取化合物11k(60.0mg,0.30mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将5a(76.0mg,0.30mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶得灰褐色固体12k(90.0mg,65.2%),m.p.80.8-81.9℃。1H NMR(400MHz,CDCl3)δ:8.53–8.28(m,2H,thiourea-H),8.05(s,1H,pyridinyl-H),7.81(s,1H,pyridinyl-H),7.71(s,2H,Ar-H),7.45(d,J=8.4Hz,1H,pyridinyl-H),7.36–7.25(m,1H,CONHCH3),7.20–7.00(m,3H,CONHCH3),6.75(dd,J=33.2,12.4Hz,1H,Ar-H),3.03(d,J=4.8Hz,1H,CONH-CH3),2.40(s,1H,Ar-CH3).13C NMR(100MHz,CDCl3)δ:180.1(1C),165.6(1C),164.5(C),154.0(1C),152.4(1C),149.7(1C),139.2(1C),135.9(1C),132.7(1C),130.1(1C),126.8(1C),126.1(1C),123.6(1C),119.6(1C)114.8(1C),110.4(1C),29.71(1C),26.21(1C),18.22(1C)。
12)N-(4-甲苯基)-N’-[4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12l)的制备
取化合物对甲基苯胺13a(36.0mg,0.34mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6b(100mg,0.34mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶黄色固体12l(110mg,82.5%),m.p.96.5-97.3℃。1H NMR(400MHz,DMSO-d6)δ:9.81(d,J=7.8Hz,2H,thiourea-H),8.80(d,J=4.2Hz,1H,pyridinyl-H),8.53(d,J=5.5Hz,1H,pyridinyl-H),7.60(d,J=8.6Hz,2H,Ar-H),7.41(s,1H,pyridinyl-H),7.34(d,J=7.9Hz,2H,Ar-H),7.18(dd,J=15.9,8.4Hz,4H,Ar-H),2.79(d,J=4.5Hz,3H,CONH-CH3),2.29(s,3H,Ar-CH3).
13)N-苯基-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12m)的制备
取化合物对甲基苯胺13a(75.0mg,0.70mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(210mg,0.70mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶白色固体12m(230mg,80.6%),m.p.101.3-102.6℃。1H NMR(400MHz,DMSO-d6)δ:9.68(s,1H,thiourea-H),9.28(s,1H,thiourea-H),8.80(d,J=4.8Hz,1H,pyridinyl-H),8.53(s,1H,pyridinyl-H),7.44(d,J=2.5Hz,1H,pyridinyl-H),7.35(dd,J=11.8,8.5Hz,3H,Ar-H),7.22–7.13(m,4H,Ar-H),7.05(dd,J 8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.28(d,J=8.5Hz,6H,Ar-CH3)。
14)N-(3-氯苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12n)的制备
取化合物13b(90.0mg,0.70mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(210mg,0.70mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得黄色固体12n(175mg,58.5%),m.p.153.3-154.6℃。1H NMR(400MHz,DMSO-d6)δ:9.90(s,1H,thiourea-H),9.56(s,1H,thiourea-H),8.81(d,J=4.8Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.71(s,1H,pyridinyl-H),7.40(m,J=16.4,5.3Hz,4H,Ar-H),7.23–7.15(m,3H,Ar-H),7.07(dd,J=8.5,2.5Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
15)N-(2,5-二氯苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12o)的制备
取化合物2,5-二氯苯胺13c(162mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~1:1体系梯度洗脱)得白色固体12o(193mg,41.8%),m.p.107.0-108.1℃。1H NMR(400MHz,DMSO-d6)δ:10.67(d,J=48.7Hz,1H,thiourea-H),9.63(s,1H,thiourea-H),8.81(d,J=4.9Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.89(dd,J=8.9.2.5Hz,1H,pyridinyl-H),7.82(d,J=2.5Hz,1H,Ar-H),7.57(d,J=8.9Hz,1H,Ar-H),7.39(s,1H,Ar-H),7.22(ddd,J=34.3,10.3,5.6Hz,3H,Ar-H),7.07(d,J=7.9Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.21(s,3H,Ar-CH3)。
16)N-(3-氰基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12p)的制备
取化合物3-氰基苯胺13d(118mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得白色固体12p(200mg,47.9%),m.p.77.1-77.9℃。1H NMR(400MHz,CDCl3)δ:8.47(d,J=5.6Hz,1H,thiourea-H),8.10(s,1H,thiourea-H),7.96(s,1H,pyridinyl-H),7.87(d,J=12.6Hz,2H,pyridinyl-H),7.79(d,J=7.7Hz,1H,Ar-H),7.66(d,J=2.3Hz,1H,Ar-H),7.51(m,J=15.4,7.7Hz,2H,Ar-H),7.40(d,J=9.5Hz,1H,Ar-H),7.15–7.08(m,2H,Ar-H),7.06–7.02(m,1H,CONHCH3),3.02(d,J=5.1Hz,3H,CONH-CH3),2.38(s,3H,Ar-CH3).13C NMR(100MHz,CDCl3)δ:180.8(1C),165.8(1C),164.7(1C),153.4(1C),152.0(1C),150.1(1C),139.3(1C),138.6(1C),132.5(1C),129.9(1C),129.7(1C),129.4(1C),128.1(1C),123.4(1C),119.4(1C),118.2(1C),115.1(1C),112.7(1C),109.9(1C),26.25(1C),18.25(1C),14.20(1C)。
17)N-(4-羟基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12q)的制备
取化合物对羟基苯胺13e(237mg,0.70mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(90.0mg,0.80mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=5:1)重结晶白色固体12q(0.18g,55.0%),m.p.189.8-190.3℃。1H NMR(400MHz,DMSO-d6)δ:9.44(d,J=26.0Hz,2H,thiourea-H),9.10(s,1H,OH),8.80(d,J=4.9Hz,1H,pyridinyl-H),8.53(d,J=5.6Hz,1H,pyridinyl-H),7.44(d,J=2.5Hz,1H,pyridinyl-H),7.35(d,J=8.5Hz,1H,Ar-H),7.20–7.11(m,4H,Ar-H),7.03(dd,J=8.5,2.7Hz,1H,CONHCH3),6.74(d,J=8.7Hz,1H),2.79(d,J=4.8Hz,3H,CONH-CH3),2.25(s,3H,Ar-CH3)。
18)N-(2-甲基-4-羟基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12r)的制备
取化合物2-甲基对羟基苯胺13f(82.0mg,0.67mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(200mg,0.67mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶白色固体12r(230mg,81.7%),m.p.197.0-197.4℃。1H NMR(400MHz,DMSO-d6)δ:9.39(s,1H,thiourea-H),9.09(s,1H,thiourea-H),8.79(d,J=4.9Hz,1H,pyridinyl-H),8.52(d,J=5.6Hz,1H,pyridinyl-H),7.43(d,J=2.4Hz,1H,pyridinyl-H),7.32(d,J=8.4Hz,1H,Ar-H),7.20–7.09(m,2H,Ar-H),7.03(dd,J=8.5,2.6Hz,1H,CONHCH3),6.97(d,J=8.4Hz,1H,Ar-H),6.66–6.57(m,2H,Ar-H),2.79(d,J=4.8Hz,3H,CONH-CH3),2.23(s,3H,Ar-CH3),2.16(s,3H,OH-Ph-CH3)。
19)N-(2-甲酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12s)的制备
取化合物2-氨基苯甲酰胺13g(91.0mg,0.67mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(200mg,0.67mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=4:1)重结晶白色固体12s(200mg,69.0%),m.p.178.2-178.7℃。1H NMR(400MHz,DMSO-d6)δ:11.35(s,1H,thiourea-H),10.02(s,1H,thiourea-H),8.86(d,J=4.9Hz,2H,pyridinyl-H),8.55(d,J=5.6Hz,1H,pyridinyl-H),8.23(s,1H,Ar-H),7.76–7.65(m,2H,CONH2),7.57(d,J=2.4Hz,1H,Ar-H),7.50–7.44(m,1H,CONHCH3),7.34(d,J=8.5Hz,1H,Ar-H),7.25–7.20(m,2H,Ar-H),7.16–7.08(m,2H,Ar-H),2.81(d,J=4.8Hz,3H,CONH-CH3),2.23(s,3H,Ar-CH3)。
20)N-(3,5-二甲氧基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12t)的制备
取化合物3,5-二甲氧基苯胺13h(153mg,0.67mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(200mg,0.67mmol)溶于无水乙醇(10.0mL)加入上述体系中,同11a反应处理方法,石油醚/乙酸乙酯(v/v=3:1)重结晶白色固体12t(420mg,92.0%),m.p.130.9-131.6℃。1H NMR(400MHz,CDCl3)δ:8.42(d,J=5.6Hz,1H,thiourea-H),8.06(d,J=4.6Hz,1H,thiourea-H),7.86(s,1H,pyridinyl-H),7.73–7.65(m,2H,pyridinyl-H),7.47(d,J=8.4Hz,1H,Ar-H),7.05–6.94(m,3H,Ar-H),6.58(d,J=1.9Hz,2H,Ar-H),6.42(d,J=2.0Hz,1H,CONHCH3),3.82(s,6H,OCH3),3.02(d,J=5.1Hz,3H,CONH-CH3),2.32(s,3H,Ar-CH3)。
21)N-(4-乙氧甲酰基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12u)的制备
取化合物4-氨基苯甲酰乙酯13i(165mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得黄色固体12u(60.0mg,13.0%),m.p.140.6-142.1℃。1H NMR(400MHz,CDCl3)δ:8.45(d,J=5.6Hz,1H,thiourea-H),8.08(d,J=8.6Hz,3H,pyridinyl-H),7.95(s,1H,thiourea-H),7.87(s,1H,Ar-H),7.69(d,J=2.4Hz,1H,Ar-H),7.59(d,J=8.6Hz,2H,Ar-H),7.41(d,J=8.5Hz,1H,Ar-H),7.09–7.05(m,2H,Ar-H),7.02(dd,J=8.5,2.6Hz,1H,CONHCH3),4.39(d,J=7.1Hz,2H,CH2CH3),3.02(d,J=5.1Hz,3H,CONH-CH3),2.35(s,3H,Ar-CH3),1.41(t,3H,CH2CH3).13C NMR(100MHz,CDCl3)δ:180.4(1C),165.9(1C),165.8(1C),164.6(1C),153.3(1C),152.1(1C),149.9(1C),141.9(1C),138.3(1C),130.6(1C),129.8(1C),127.8(1C),123.4(1C),119.3(1C),114.9(1C),110.1(1C),61.09(1C),26.22(1C),18.26(1C),14.33(1C)。
22)N-(3-三氟甲基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12v)的制备
取化合物间三氟甲基苯胺13j(161mg,1.0mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.0mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得黄色固体12v(140mg,30.4%),m.p.77.8-78.9℃。1H NMR(400MHz,DMSO-d6)δ:9.99(s,1H,thiourea-H),9.65(s,1H,thiourea-H),8.81(d,J=4.8Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.96(s,1H,pyridinyl-H),7.79(d,J=8.2Hz,1H,Ar-H),7.57(t,J=7.9Hz,1H,Ar-H),7.51–7.36(m,3H,Ar-H),7.22–7.16(m,2H,Ar-H),7.08(dd,J=8.5,2.7Hz,1H,CONHCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.28(s,3H,Ar-CH3)。
23)N-(4-三氟甲基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12w)的制备
取化合物对三氟甲基苯胺13k(161mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:1体系梯度洗脱)得白色固体12w(90.0mg,33.2%),m.p.127.4-128.9℃。1H NMR(400MHz,CDCl3)δ:8.42(s,1H,thiourea-H),8.08(s,1H,thiourea-H),7.82(s,1H,pyridinyl-H),7.65(s,1H,pyridinyl-H),7.49(s,2H,Ar-H),7.15–6.87(m,4H,Ar-H),6.14(q,1H,CONHCH3),5.65(d,J=28.1Hz,1H,Ar-H),3.01(s,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
24)N-(4-氯-3-三氟甲基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12x)的制备
取化合物对氯间三氟甲基苯胺13l(196mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~1:2体系梯度洗脱)得白色固体12x(90.0mg,36.4%),m.p.158.9-159.4℃。1H NMR(400MHz,DMSO-d6)δ:10.04(s,1H,thiourea-H),9.71(s,1H,thiourea-H),8.81(d,J=4.9Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),8.10(d,J=2.4Hz,1H,pyridinyl-H),7.84(dd,J=8.7,2.4Hz,1H,Ar-H),7.68(d,J=8.7Hz,1H,Ar-H),7.42(dd,J=21.7,5.5Hz,2H,Ar-H),7.22–7.15(m,2H,Ar-H),7.08(dd,J=8.5,2.8Hz,1H,CONHCH3),2.79(d,J=4.9Hz,3H,CONH-CH3),2.27(s,3H,Ar-CH3)。
25)N-(3-硝基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12y)的制备
取化合物间硝基苯胺13m(138mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~1:2体系梯度洗脱)得黄绿色固体12y(180mg,41.0%),m.p.142.8-143.1℃。1H NMR(400MHz,DMSO-d6)δ:10.14(s,1H,thiourea-H),9.72(s,1H,thiourea-H),8.80(q,J=4.5Hz,1H,pyridinyl-H),8.66–8.51(m,2H,pyridinyl-H),8.01–7.91(m,2H,Ar-H),7.62(t,J=8.2Hz,1H,Ar-H),7.42(dd,J=17.5,5.5Hz,2H,Ar-H),7.25–7.16(m,2H,Ar-H),7.09(dd,J=8.4,2.7Hz,1H,CONHCH3),2.80(d,J=4.9Hz,3H,CONH-CH3),2.29(s,3H,Ar-CH3)。
26)N-(2-邻甲基-3-硝基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12z)的制备
取化合物邻甲基间硝基苯胺13n(152mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~1:2体系梯度洗脱)得黄色固体12z(180mg,26.6%),m.p.115.2-116.6℃。1H NMR(400MHz,DMSO-d6)δ:9.51(d,J=12.5Hz,1H,thiourea-H),8.79(d,J=4.7Hz,1H,thiourea-H),8.53(d,J=5.7Hz,1H,pyridinyl-H),7.83(d,J=7.9Hz,1H,pyridinyl-H),7.59(d,J=7.7Hz,1H,pyridinyl-H),7.46-7.38(m,1H,Ar-H),7.17(dd,J=5.3,2.Hz,1H,CONHCH3),7.11–7.05(m,1H,Ar-H),6.06(s,1H,Ar-H),2.79(d,J=4.7Hz,3H,CONH-CH3),2.51(s,3H,CF3Ph-CH3),2.31(d,J=18.9Hz,3H,Ar-CH3)。
27)N-(2-甲基-5-硝基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12aa)的制备
取化合物邻甲基-5-硝基苯胺13o(152mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~2:1体系梯度洗脱)得黄绿色固体12aa(230mg,51.0%),m.p.96.4-97.4℃。1H NMR(400MHz,DMSO-d6)δ:9.64(s,1H,thiourea-H),9.45(s,1H,thiourea-H),8.80(d,J=4.7Hz,1H,thiourea-H),8.53(d,J=5.6Hz,1H,pyridinyl-H),8.20(s,1H,pyridinyl-H),8.05(d,J=8.3Hz,1H,pyridinyl-H),7.55(d,J=8.5Hz,1H,Ar-H),7.44(dd,J=11.0,5.2Hz,2H,Ar-H),7.18(s,1H,Ar-H),7.09(d,J=8.2Hz,3H,Ar-H),2.79(d,J=4.6Hz,3H,Ar-CH3),2.38(s,1H,CONH-CH3),2.30(s,3H,Ar-CH3)。
28)N-(3-甲磺酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ab)的制备
取化合物9a(186mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,析出大量固体,过滤,石油醚/乙酸乙酯(v/v=3:1)重结晶得白色固体12ab(450mg,92.7%),m.p.174.0-175.1℃。1H NMR(400MHz,DMSO-d6)δ:9.84(s,1H,thiourea-H),9.39(s,1H,thiourea-H),8.79(s,1H,pyridinyl-H),8.52(d,J=5.3Hz,1H,pyridinyl-H),7.94–6.45(m,7H,Ar-H),3.03(d,J=6.9Hz,3H,CH3SO2NH),2.77(s,3H,CONH-CH3),2.26(s,3H,Ar-CH3)。
29)N-(4-甲磺酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ac)的制备
取化合物9b(186mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,析出大量固体,过滤,石油醚/乙酸乙酯(v/v=3:1)重结晶得白色固体12ac(240mg,49.4%),m.p.144.9-145.1℃。1H NMR(400MHz,DMSO-d6)δ:9.70(s,2H,thiourea-H),9.29(s,1H,pyridinyl-H),8.80(s,1H,pyridinyl-H),8.54(s,1H,pyridinyl-H),7.42(s,4H,Ar-H),7.12(d,J=53.7Hz,3H,Ar-H),7.05(s,1H,CH3SO2NH),2.98(s,3H,CH3SO2NH),2.79(s,3H,CONH-CH3),2.26(s,3H,Ar-CH3)。
30)N-(4-甲基-3-甲磺酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ad)的制备
取化合物9c(200mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=4:1~2:1体系梯度洗脱)得白色固体12ad(12.0mg,24.0%),m.p.133.5-135.3℃。1H NMR(400MHz,CDCl3)δ:8.49–8.42(m,1H,thiourea-H),8.13–8.07(m,1H,thiourea-H),7.84–7.68(m,3H,pyridinyl-H),7.47–7.22(m,3H,Ar-H),7.09(s,3H,Ar-H),3.54(d,J=10.3Hz,3H,CH3SO2NH),3.13–2.90(m,3H,CONH-CH3),2.51–2.03(m,6H,Ar-CH3)。31)N-(3-甲基-4-甲磺酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ae)的制备
取化合物9d(200mg,1.00mmol)溶解于无水乙醇(5.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,析出大量固体,过滤,石油醚/乙酸乙酯(v/v=1:1)重结晶得白色固体12ae(440mg,86.1%),m.p.163.6-164.4℃。1H NMR(400MHz,Acetone-d6)δ:9.15(s,1H,thiourea-H),8.87(s,1H,thiourea-H),8.49(d,J=5.6Hz,1H,pyridinyl-H),8.16(s,1H,pyridinyl-H),7.57(d,J=2.5Hz,1H,pyridinyl-H),7.44(dd,J=9.0,5.4Hz,3H,Ar-H),7.39(d,J=8.3Hz,1H,Ar-H),7.13–7.10(m,2H,Ar-H),7.05(q,1H,CONH-CH3),3.01(s,3H,CH3SO2NH),2.94(s,3H,CONH-CH3),2.40(s,3H,Ar-CH3),2.37(s,3H,Ar-CH3)。
32)N-(3-甲氧基-4-甲磺酰胺苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12af)的制备
取化合物9e(216mg,1.00mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得白色固体12af(0.23g,44.8%),m.p.121.0-122.3℃。1H NMR(400MHz,DMSO-d6)δ:9.85(s,1H,thiourea-H),9.36(s,1H,thiourea-H),9.14–8.69(m,2H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),7.45–7.31(m,3H,Ar-H),7.20(dd,J=8.3,4.1Hz,2H,Ar-H),7.15(s,1H,CH3SO2NH),7.06(dd,J=8.5,2.6Hz,1H,Ar-H),7.01(dd,J=8.5,2.0Hz,1H,CONH-CH3),3.82(s,3H,CH3SO2NH),2.93(s,3H,OCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.28(s,3H,Ar-CH3)。
33)N-(3-吡啶基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ag)的制备
取化合物3-氨基吡啶13p(94.0mg,1.00mmol)溶解于无水乙醇(10.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=2:1~1:2体系梯度洗脱)得黄色固体12ag(100mg,25.4%),m.p.110.4-111.8℃。1H NMR(400MHz,DMSO-d6)δ:9.90(s,1H,thiourea-H),9.70(s,1H,thiourea-H),8.78(s,1H,pyridinyl-H),8.58(d,J=29.9Hz,2H,pyridinyl-H),8.33(s,1H,pyridinyl-H),7.95(s,1H,pyridinyl-H),7.40(d,J=3.3Hz,3H,Ar-H),7.17(s,2H,pyridinyl-H),7.07(d,J=6.6Hz,1H,CONH-CH3),2.80(s,3H,CONH-CH3),2.28(s,3H,Ar-CH3)。
34)N-(2-甲氧基-5-苯胺甲酰基苯基)-N’-[2-甲基-4-[2-(N-甲基氨甲酰基)-4-吡啶基氧基]苯基]硫脲(12ah)的制备
取化合物4-甲氧基-3-氨基苯酰苯胺13q(240mg,1.00mmol)溶解于无水乙醇(20.0mL)置于100mL圆底烧瓶中,将6a(300mg,1.00mmol)溶于无水乙醇(15.0mL)加入上述体系中,同11a反应处理方法,柱层析分离(石油醚/乙酸乙酯v/v=1:1~1:4体系梯度洗脱)得黄色固体12ah(32.0mg,5.9%),m.p.170.8-172.0℃。1H NMR(400MHz,DMSO-d6)δ:10.14(s,1H,CONHPh),9.46(s,1H,thiourea-H),9.30(s,1H,thiourea-H),8.81(d,J=4.8Hz,1H,pyridinyl-H),8.54(d,J=5.6Hz,1H,pyridinyl-H),8.37(s,1H,pyridinyl-H),7.90(dd,J=8.6,1.9Hz,1H,Ar-H),7.78(d,J=7.9Hz,2H,Ar-H),7.46–7.29(m,4H,Ar-H),7.25–7.10(m,4H,Ar-H),7.08–7.04(m,1H,CONH-CH3),3.93(s,3H,OCH3),2.79(d,J=4.8Hz,3H,CONH-CH3),2.29(s,3H,Ar-CH3)。
二、抗氧化活性测试
测定化合物的抗氧化活性采用DPPH通法,具体参见专利CN 2016100321177中的公开描述。根据目标化合物测定的吸光度值,用统计软件计算各自IC50值,将化合物的抗氧化活性的测试结果分析如下(表1)。
表1.目标化合物抗氧化活性测试结果
*由于清除率小于50%,未测其IC50。
如上表所示,目标化合物的清除率几乎都超过了50%,表现出了不同的抗氧化活性。34个目标化合物中,清除率较高的有12q,12ae,12r清除率都达到90%。活性最好的化合物12q对DPPH的清除活性IC50为0.025mg/mL,活性与阳性对照的抗坏血酸相近,甲磺酰胺基产物活性也较高,12ae为0.12mg/mL。在化学中,自由基夺取电子形成稳定化合物的过程称为“氧化”,酚羟基易形成自由基与DPPH形成共价化合物而表现出很强的抗氧化活性。化合物12j和12r的浓度对DPPH清除率曲线如图3中,(a)和(b)所示。
三、抗病毒活性
目标化合物对疱疹病毒、牛痘病毒、呼吸道肠道病毒、Coxsackie病毒、Feline冠状疱疹病毒、HIV病毒等都有一定的抑制活性。测定原理和通法参见专利CN 2016100321177中的公开描述。
a)测定目标化合物12a-12ah对病毒感染的HEL细胞株的细胞毒性及抗病毒活性测试,结果如表2所示:
表2.HEL细胞株抗病毒活性测试结果
如表2所示目标化合物细胞毒性普遍较小,其中12x的毒性最大,病毒抑制活性也较差。其中化合物12h,12j,12y,12ag具有一定的抑制单纯疱疹病毒-1(KOS)、单纯疱疹病毒-2(G)活性,并且细胞毒性较弱。其余化合物在测试浓度下未表现出抗病毒活性。
b)目标化合物HeLa细胞株的细胞毒性及抗病毒活性测试
目标化合物HeLa细胞株的细胞毒性及抗病毒活性测试结果如下表3所示:
表3.目标化合物HeLa细胞株的细胞毒性及抗病毒活性
如表3所示,目标化合物对HeLa细胞株毒性差距很大,有很多比阳性药安全的比如12a、12b、12i等,但是目标化合物在测试浓度下未表现出抑制活性,EC50的数值都要大于测试浓度。
c)目标化合物Vero细胞株细胞毒性及抗病毒活性
目标化合物Vero细胞株细胞毒性及抗病毒活性如下表4所示:
表4.目标化合物Vero细胞株细胞毒性及抗病毒活性
如表4所示,目标化合物对上述病毒在测试浓度下未表现出抑制活性,个别化合物表现出极高的细胞毒性,如12v,12x,12w。
d)目标化合物CRFK细胞株抗猫科冠状、疱疹病毒活性测试
如以下表5所示:
表5.目标化合物CRFK细胞株抗猫科冠状、疱疹病毒活性
如上表5所示,目标化合物抗猫科冠状、疱疹病毒活性普遍较弱,细胞毒性较阳性对照药较小,12v,12w和12x的细胞毒性最强。
e)对于流感病毒采用visual CPE score和MTS法测定目标化合物对甲型流感病毒H1N1、H3N2和乙型流感病毒的抑制活性,如下表6所示
表6.目标化合物在MDCK细胞株细胞毒性及抗流感病毒活性
如上表6所示,目标化合物抗A型流感病毒H1N1,H3N2,B型流感病毒较阳性对照药弱,在测试浓度下目标化合物未表现出抑制活性。12f,12v,12x的细胞毒性都要比阳性对照药大。
综上所有表格所示,分析总结得到抗病毒活性较高的部分化合物12h,12j,12y,12ag,它们对单纯疱疹病毒(Herpes simplex virus-1(KOS))和单纯疱疹病毒(Herpes simplexvirus-2(G))具有一定抑制活性,抑制活性及最小细胞毒浓度(MCC)与阳性对照药溴夫定(Brivudin)、西多福韦(Cidofovir)、阿昔洛韦(Acyclovir)和更昔洛韦(Ganciclovir)如下表7所示,而对牛痘病毒(Vaccinia virus)、疱疹性口炎病毒(Vesicular stomatitis virus)、单形疱疹病毒(Herpes simplex virus-1(TK-KOS ACVr))、柯萨奇病毒(Coxsackie VirusB4)、甲型流感病毒H1N1(Influenza A H1N1)、甲型流感病毒H3N2(Influenza A H3N2)在测试浓度下没有抑制作用。
表7.部分目标化合物抗病毒活性总结
由上表7可以看出,吡啶取代的硫脲衍生物的抑制活性要比苯基取代的要高。A环为硝基苯取代的硫脲衍生物活性较高,但比吡啶取代物活性要差。
2、抗HIV活性
对目标化合物12a-12ac进行抗HIV活性测试内容包括对HIV-1野生型病毒株(IIIB),HIV-2病毒株(ROD)的抑制活性,结果用IC50表示。同时进行了采用MTT法对细胞毒性测试,结果用CC50表示。测定原理和通法参见专利CN 2016100321177中的公开描述。抗HIV活性测试结果列在下列表中。
表8.目标化合物抗HIV活性
如上表8所示,目标化合物的毒性CC50较阳性对照药较大,选择指数(SI)较小。
综上所述,本发明公开的硫脲类衍生物是一系列同时含有各种取代芳环结构和不对称取代硫脲结构的化合物,均未见文献报道。具体合成方法如下:2-吡啶甲酸(1)中72℃下在氯化亚砜中反应生成4-氯吡啶-2-甲酰氯(2);中间体2与甲胺水溶液在低温条件下反应,经氢氧化钠溶液碱化生成N-甲基-4-氯吡啶-2-甲酰胺(3);中间体3和对氨基苯酚(4)在碳酸铯存在下,105℃发生威廉姆森成醚反应,反应生成4-(4-氨基苯氧基)-N-甲基-2-吡啶甲酰胺(5)。对于苯环上强吸电子基取代的苯胺,在二硫化碳、三乙胺、氯甲酸甲酯作用下不能生成相应的异硫氰酸酯,或者产率很低时,只有将中间体5合成异硫氰酸酯再与不同取代苯胺缩合生成12。中间体5在二硫化碳、三乙胺作用下生成其三乙胺盐,在硫酸亚铁催化下转化为化合物N-甲基-4-(3-R1-4-异硫氰酸酯苯氧基)吡啶-2-甲酰胺(6)。对于苯环上不被取代或者斥电子基取代的苯胺,则采用常用芳胺、二硫化碳和叔胺(如三乙胺、吡啶)反应先生成二硫代氨基甲酸盐,然后在氯甲酸甲酯作用下生成异硫氰酸酯的方法。含斥电子基的苯胺13,在二硫化碳、三乙胺作用下生成三乙胺盐中间体,最终在氯甲酸甲酯转化下生成取代苯异硫氰酸酯(11)。分别将中间体6和中间体9,中间体5和中间体11,化合物13和中间体6溶解于一定量的无水乙醇中,室温搅拌,直接反应得12系列目标化合物,为不同取代芳基-N’-[2-R1-4-[2-(N-甲基胺基甲酰基)-4-吡啶基氧基]苯基]硫脲。对于中间体9的制备,可采用苯环不同位置硝基取代苯胺7,在二氯甲烷作溶剂,吡啶这一弱碱作用下与甲磺酰氯反应,生成化合物苯甲磺酰胺8。8溶于乙醇在氯化亚锡作为还原剂的作用下,硝基还原成氨基(如图2所示)。苯胺室温条件下,再与中间体6缩合得目标产物不同取代的氨基苯甲磺酰胺(9)。实验证明,只有当7上硝基和氨基处于间位或对位时,才能与6发生缩合反应。所有新型中间体及34个目标化合物经熔点,13C NMR、MS以及1H NMR等进行鉴定。
对所有目标化合物的生物活性测试分析包括DPPH抗氧化活性、抗病毒活性测定。结果表明,多数目标化合物都具有较好的抗氧化活性,表现出对DPPH具有较高清除率。所有目标化合物的DPPH清除率几乎都超过了50%,表现出不同的抗氧化活性。清除率较高的化合物有12q,12ae和12r清除率都达到90%。其中化合物12q抗氧化活性活性较好,对DPPH的清除活性IC50为0.025mg/mL,活性与阳性对照的抗坏血酸相近,甲磺酰胺基产物活性也较高,12ae的IC50为0.12mg/mL。化合物12h,12j,12y和12ag具有一定的抗疱疹病毒活性,可抑制单纯疱疹病毒-1(KOS)、单纯疱疹病毒-2(G)活性,且细胞毒性较弱。分析上述化合物的构效关系可知:吡啶取代的硫脲衍生物的抑制活性要比苯基取代的要高。A环为硝基苯取代的硫脲衍生物活性较高,但比吡啶取代物活性要差。总之,本专利所设计合成的化合物结构新颖,其抗氧化活性和抗病毒活性首次被揭示。另外有望还有未知的生物活性未充分阐明,可为进一步新药研发提供一定物质基础。
- 还没有人留言评论。精彩留言会获得点赞!