一种从发酵液中提取ε-聚赖氨酸及其盐酸盐的方法与流程
本发明涉及一种从发酵液中提取ε-聚赖氨酸及其盐酸盐的方法,属于生物分离工程领域。
背景技术:
ε-聚赖氨酸(ε-PL)是由链霉菌、丝状真菌或芽孢杆菌等微生物胞外分泌产生的一种同型氨基酸聚合物。它一般由25-35个L-赖氨酸单体通过α-COOH和ε-NH2脱水缩合而成,分子量通常为2500-4500Da。由于ε-PL具有水溶性好、热稳定性强、抑菌谱广等特点,目前主要作为生物食品防腐剂广泛应用于日本、韩国、欧美等国家和地区的食品工业。2014年,我国也批准了ε-PL及其盐酸盐在果蔬、米面制品、肉制品、调味品、饮料和焙烤等食品领域中的应用。事实上,ε-PL与已在食品工业中被普遍使用的生物食品防腐剂——乳酸链球菌素和纳他霉素在抑菌谱和使用范围上存在明显的互补性。因此,大力开发和推广ε-PL及其盐在食品工业中的应用,对于解决当前化学食品防腐剂引发的食品安全问题具有重要意义。
ε-PL及其盐酸盐的制备完全是依靠从链霉菌发酵液中进行分离、提取和精制,其分离提取成本占到总生产成本的70-80%。可见,分离提取技术对于实现低成本ε-PL的生物制造具有重要贡献。然而,目前已公布的ε-PL分离提取方法,主要集中于单元提取技术的研究与开发,如菌体的固液分离、离子交换吸附与洗脱、产品脱色等;只有少量报道涉及ε-PL较为完整的提取工艺路线。
事实上,不同的物料特性决定着采用不同的分离提取方法。例如,对于发酵液中仅含有20-30g/Lε-PL和30g/L菌体干重的发酵液(Bioresource Technology,2015,187:70–76;ZL200910069817.5),采用专利ZL200910152931.2和ZL201110053004.2能够实现纯度为95-97%的ε-PL收率达到70-80%。然而,将上述两种方法应用于发明人团队公布专利201510021744.6制备的发酵液(54.7g/Lε-PL和76.35g/L菌体干重),却无法实现菌体的有效分离,且制备的ε-PL纯度仅为60%左右,收率也只有50-60%。可见,已公布ε-PL分离提取方法仅适用于低ε-PL浓度和低菌体量的发酵液体系,而不适用于高ε-PL浓度和高菌体量的发酵液中分离提取ε-PL。这主要是因为高ε-PL浓度的发酵液中往往含有更高的菌体量、更多的代谢副产物和更复杂的料液性质。然而,高ε-PL浓度的发酵方式是提高发酵效率和降低生产成本的主要手段。因此,研究和开发从高ε-PL浓度和高菌体量的发酵液中分离提取ε-PL是实现低成本和高效率ε-PL生物制造的根本任务。
技术实现要素:
为了解决上述问题,本发明提供了一种从发酵液中高效提取ε-PL及其盐酸盐的方法。
为实现上述目的,本发明的从发酵液中提取ε-PL及其盐酸盐的方法,包括如下步骤:
(1)发酵液固液分离:发酵液经过絮凝或稀释后,通过过滤或离心去除菌丝体和水不溶性杂质,获得发酵液清液;
(2)超滤:利用超滤膜系统,将发酵液清液中的水溶性大分子杂质和水不溶性杂质去除,得到超滤透过液;
(3)离子交换:将超滤透过液压入一级离子交换柱中进行吸附,直至树脂吸附饱和;随后用洗涤剂进行除杂,再用洗脱剂对树脂进行解吸附;收集获得的洗脱液,再压入二级离子交换柱中,收集获得流出液;
(4)纳滤:利用纳滤膜系统,将步骤(3)获得的流出液进行脱盐和浓缩,收集纳滤浓缩液;
(5)脱色:将收集的纳滤浓缩液进行脱色,获得脱色液;
(6)浓缩:将步骤(5)获得的脱色液,浓缩至ε-聚赖氨酸含量为10~30%;
(7)干燥:将步骤(6)获得的浓缩液进行干燥处理,得到ε-聚赖氨酸及其盐酸盐成品
在本发明的一种实施方式中,所述发酵液是利用小白链霉菌CGMCC NO.10480发酵得到的。
在本发明的一种实施方式中,所述发酵液中含有50g/L以上的ε-PL和60g/L以上的菌体干重。
在本发明的一种实施方式中,所述发酵液是按照CN 201510021744.6的方法制备得到的。
在本发明的一种实施方式中,所述发酵液的制备是:将小白链霉菌CGMCC NO.10480的种子液按照6%~8%的接种量接种到发酵培养基中进行发酵,发酵过程中控制搅拌转速200~800rpm、温度28~32℃、通气量0.5~2vvm、溶氧28~32%;同时,在发酵过程中,当pH首次自发降为4.5~5.2时开始,保持发酵pH值4.8~5.2并维持10~15h,再将pH值降为2.8~3.2并维持22~26h,后将pH上调到4.2~4.8并维持至发酵结束;当发酵液中残留的甘油或葡萄糖浓度降为10g/L以下时,流加甘油或者葡萄糖溶液使其在发酵液中的浓度控制在9.5~10.5g/L;当发酵液中NH4+-N浓度降到1g/L以下时,流加硫酸铵溶液,使NH4+-N浓度维持在0.8~1.2g/L。
在本发明的一种实施方式中,发酵培养基配方(g/L):葡萄糖或甘油83,(NH4)2SO48,鱼粉15,玉米浆5,KH2PO45,MgSO4·7H2O 2,FeSO4·7H2O 0.1,pH 6.8。
在本发明的一种实施方式中,步骤(1)中,所述的发酵液絮凝,可以是热絮凝,也可以是絮凝剂介导的絮凝。
在本发明的一种实施方式中,步骤(1)中,所述的过滤,可以是板框过滤,也可以是膜过滤。
在本发明的一种实施方式中,步骤(1)中,所述的离心,可以是碟式离心机离心、沉降式离心机离心,也可以是沉降过滤式离心机离心。
在本发明的一种实施方式中,步骤(2)中,所述超滤膜系统,采用的超滤膜元件可以是管式膜,也可以是卷式膜;超滤膜的截留分子量为10~1000KDa。
在本发明的一种实施方式中,步骤(2)具体是:将发酵液清液经过截留分子量为10~1000KDa的超滤膜,在操作压力0.1~0.15MPa条件下浓缩10~20倍。
在本发明的一种实施方式中,步骤(3)中,超滤透过液调节pH至5.0-8.5再压入一级离子交换柱中进行吸附。
在本发明的一种实施方式中,步骤(3)中,所述的一级离子交换柱中装填的是弱酸或强酸型阳离子交换树脂;树脂的活性交换基团可以是H+,也可以是Na+或NH4+;
在本发明的一种实施方式中,步骤(3)中,所述的洗涤剂可以是去离子水,也可以是稀酸或稀碱水溶液;洗脱剂可以是盐酸水溶液,也可以是NaOH水溶液或氨水溶液。
在本发明的一种实施方式中,步骤(3)中,所述的二级离子交换柱中装填的是弱碱或强碱型阴离子交换树脂。
在本发明的一种实施方式中,步骤(4)中,所述纳滤膜系统采用的纳滤膜元件是卷式膜,纳滤膜的截留分子量为0.1-1.0KDa;
在本发明的一种实施方式中,步骤(4)纳滤的操作压力为1.0~1.5MPa。
在本发明的一种实施方式中,步骤(4)中,所述脱盐方式可以是间歇恒容式、连续恒容式或一次性添加的一种或两种。
在本发明的一种实施方式中,步骤(4)中,所述浓缩实现ε-PL及其盐浓度至5-10%。
在本发明的一种实施方式中,步骤(5)的脱色介质可以是大孔树脂,也可以是活性炭。
本发明的有益效果:
(1)本发明采用超滤对离子交换的上样液进行深度预处理,这一方面使得上样液澄清度提高,减轻了树脂的污染并大大延长树脂的使用寿命;另一方面,也去除了上样液中的大分子杂质(如蛋白质、核酸等),有效的提升了树脂的吸附量和使用效率;
(2)本发明采用两级离子交换对目标产品ε-PL进行正吸附和负吸附,以最大程度地去 除杂质,提高离子交换树脂提纯的效果。
(3)本发明采用纳滤方式进行脱盐,可以在最大程度上去除分离提取过程中因反复调节pH值而引入的盐分,确保成品的灰分控制在合理水平,显著提升了成品纯度,这是ε-PL提取工艺中首次引入脱盐操作单元。
(4)本发明将膜分离技术(超滤和纳滤)引入到ε-PL分离提取工艺,具有自动化程度高、运行费用低、料液无二次污染且能耗低、污染小等优势。
(5)按照本发明的提取方法,所得的ε-PL及其盐酸盐产品的回收率不低于70%,纯度不低于95%。
生物材料保藏
小白链霉菌(Streptomyces albulus),分类学命名为Streptomyces albulus,已于2015年1月30日保藏于中国普通微生物菌种保藏管理中心,保藏编号为CGMCC No.10480,保藏地址为北京市朝阳区北辰西路1号院3号。该菌株已于2016年6月15日在Appl Biochem Biotechnol杂志的《Genome Shuffling and Gentamicin-Resistance to Improveε-Poly-L-Lysine Productivity of Streptomyces albulus W-156》一文中公开。
附图说明
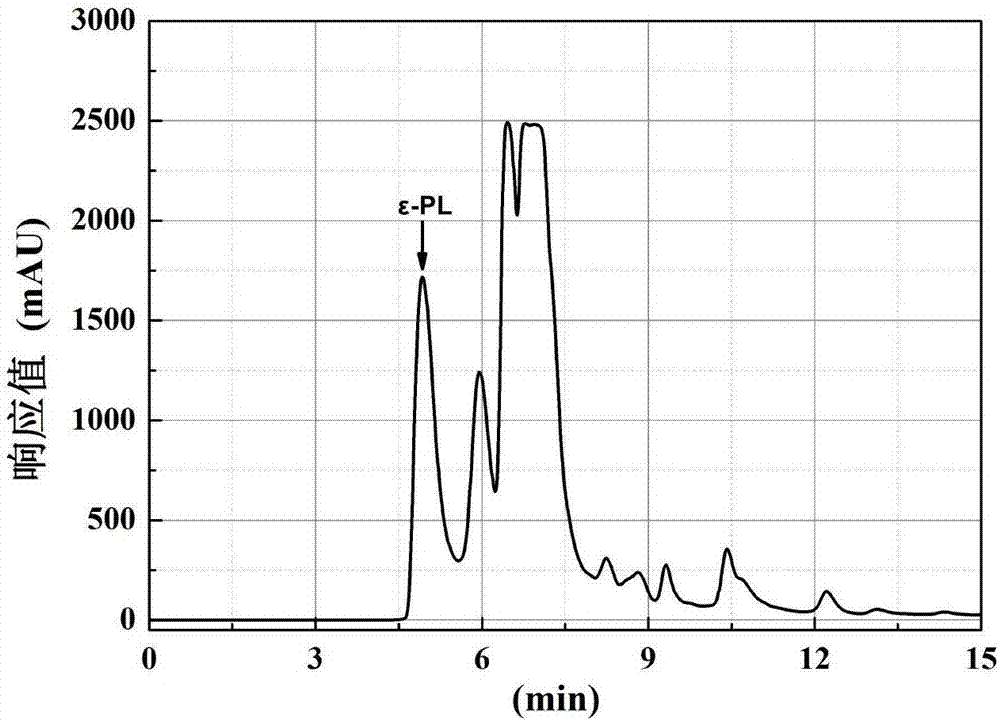
图1:发酵液高压液相色谱图;
图2:通过本发明方法最终得到的ε-PL高压液相色谱图;
图3:通过本发明方法最终得到的ε-PL盐酸盐质谱图。
具体实施方式
根据下述实施例和对照例,可以更好地理解本发明。然而,本领域的技术人员容易理解,实施例和对照例所描述的具体的物料配比、工艺条件及其结果仅用于说明本发明,而不应当也不会限制 权利要求书中所详细描述的本发明。
本发明方法中使用的离子交换树脂经过本领域技术人员公知的酸洗、碱洗和酸洗的过程,可重复使用。另外,本发明中使用的超滤膜和纳滤膜同样经过本领域技术人员公知的酸洗、碱洗和清洗剂洗的过程,可重复使用。
实施例1:ε-PL发酵液的制备
将小白链霉菌CGMCC NO.10480的种子液,按照6%的接种量接种到装有发酵培养基的5L发酵罐中,利用氨水或NaOH溶液将培养基pH值调节至7.5,开始发酵。在发酵过程中,温度控制在30℃左右,搅拌转速控制为200-800rpm,通气量为0.5-2vvm,溶氧浓度控制在30%左右;当pH值自发下降为5.0时,自动流加氨水或NaOH溶液将pH值控制在5.0左右, 保持10h;随后将pH值下调至3.0左右,并维持24h左右,后将pH值上调到4.5左右并维持至发酵结束。当发酵液中残留的甘油或葡萄糖浓度下降为10g/L时,自动流加灭菌后的纯甘油或500g/L的葡萄糖溶液,使其在发酵液中的浓度控制在10g/L左右;当发酵液中NH4+-N浓度降到1g/L时,自动流加硫酸铵溶液,使其浓度维持在1g/L。
按照上述发酵控制方法,经过192h补料分批发酵,ε-PL产量可以达到55.4g/L,菌体量为75.3g/L,湿菌体所占发酵液体积百分比为40%左右。
实施例2:ε-PL发酵液的制备
将小白链霉菌CGMCC NO.10480的种子液,按照7%的接种量接种到装有发酵培养基的5L发酵罐中,调节pH至7.0,并开始发酵。发酵过程中,控制搅拌转速200~800rpm、温度32℃、通气量0.5vvm、溶氧28%左右;当pH自发降为4.8时,添加pH调节剂将pH维持在4.8保持12h,再将pH值降为3.2并维持26h,后将pH上调到4.8并维持至发酵结束;当发酵液中残留的甘油或葡萄糖浓度降为10g/L时,流加甘油或者葡萄糖溶液使其在发酵液中的浓度控制在9.5g/L;当发酵液中NH4+-N浓度降到1g/L时,流加硫酸铵溶液,使其浓度维持在1.2g/L。
按照上述发酵控制方法,经过192h补料-分批发酵,ε-PL产量可以达到60g/L,菌体量为85g/L,湿菌体所占发酵液体积百分比为50%左右。
实施例3:ε-PL发酵液的制备
将小白链霉菌CGMCC NO.10480的种子液,按照8%的接种量接种到装有发酵培养基的5L发酵罐中,调节pH至6.5,并开始发酵。发酵过程中,搅拌转速200~800rpm、温度28℃、通气量2vvm、溶氧32%左右;在发酵过程中,当pH自发降为4.5时,添加pH调节剂将pH维持在4.5保持15h,再将pH值降为2.8并维持22h,后将pH上调到4.2并维持至发酵结束;当发酵液中残留的甘油或葡萄糖浓度降为10g/L时,流加甘油或者葡萄糖溶液使其在发酵液中的浓度控制在10.5g/L;当发酵液中NH4+-N浓度降到1g/L时,流加硫酸铵溶液,使NH4+-N浓度维持在0.8g/L。
按照上述发酵控制方法,经过192h补料-分批发酵,ε-PL产量可以达到68.7g/L,菌体量为63.8g/L,湿菌体所占发酵液体积百分比为43%左右。
实施例4:ε-PL盐酸盐的提取
按照以下方法,提取实施例1的发酵液中的ε-PL盐酸盐:
(1)在发酵液储罐中,加入发酵液4倍体积的自来水对实施例1得到的发酵液进行稀释,在室温条件下,利用碟式离心机对稀释后的发酵液进行离心,获得发酵液离心清液;
(2)将离心清液经过截留分子量为1000KDa的超滤膜,在操作压力0.3MPa条件下浓缩10倍,收集透过液;
(3)利用6M NaOH将透过液的pH值调节至5.0,然后以1BV/h的速率压入装填有强酸阳离子树脂的一级离子交换柱中进行离子交换;当树脂吸附饱和后,利用去离子水以1BV/h的速率进行洗涤;洗涤结束后,利用1M盐酸以1BV/h进行洗脱,收集洗脱液。将洗脱液以1BV/h的速率压入装填有强碱阴离子树脂的二级离子交换柱中进行负吸附,并收集流出液;
(4)利用6M盐酸将流出液pH值调节至5.0左右进行纳滤操作,纳滤膜截留分子量为100Da,操作压力为1.0MPa,并采用间歇恒容方式补加去离子水,以除去流出液中大量盐分。待脱盐结束后,再对浓缩液进行4倍浓缩;
(5)将步骤(4)脱盐后的浓缩液加热至80℃,并按质量体积比加入约1%活性炭,在搅拌条件下脱色20min后,经过滤去除活性炭,获得脱色液;
(6)将步骤(5)得到的脱色液进行蒸发浓缩,并将ε-PL浓度浓缩至10%左右;
(7)将步骤(6)得到的浓缩液,进行喷雾干燥,得到ε-PL盐酸盐产品。
按上述提取流程,所得产品中ε-PL和ε-PL盐酸盐的总含量为95.12%,即ε-PL及其盐酸盐的纯度为95.12%。采用本实施例方法得到的ε-PL及其盐酸盐的收率为72.3%。
采用相同的方法,对实施例2、3得到的发酵液进行提取,所得ε-PL及其盐酸盐纯度分别达到96.15%和95.38%,收率分别达到71.2%和70.6%。
实施例5:ε-PL的提取
(1)在发酵液储罐中,加入一定浓度絮凝剂对实施例1得到的发酵液中菌体细胞进行絮凝,在室温条件下,利用板框过滤机对稀释后的发酵液进行过滤,获得发酵液过滤清液。
(2)将过滤清液经过截留分子量为10KDa的超滤膜,在操作压力0.15MPa条件下浓缩5倍,收集透过液。
(3)利用6M NaOH将透过液的pH值调节至8.5,然后以10BV/h的速率压入装填有大孔弱酸阳离子树脂的一级离子交换柱中进行离子交换;当树脂吸附饱和后,利用去离子水以10BV/h的速率进行洗涤;洗涤结束后,利用1M NaOH以10BV/h进行洗脱,收集洗脱液。将洗脱液以10BV/h的速率压入装填有大孔弱碱阴离子树脂的二级离子交换柱中进行负吸附,并收集流出液。
(4)利用6M盐酸将流出液pH值调节至8.0左右进行纳滤操作,纳滤膜截留分子量为1000Da,操作压力为1.5MPa,并采用连续恒容方式补加去离子水,以除去流出液中大量盐 分。待脱盐结束后,再对浓缩液进行2倍浓缩;
(5)将步骤(4)脱盐后的浓缩液加热至100℃,并按质量体积比加入约1.5%活性炭,在搅拌条件下脱色30min后,经过滤去除活性炭,获得脱色液;
(6)将步骤(5)得到的脱色液进行蒸发浓缩,并将ε-PL浓度浓缩至30%左右;
(7)将步骤(6)得到的浓缩液,进行喷雾干燥,得到ε-PL产品。
按上述提取流程,所得ε-PL及其盐酸盐的纯度为96.82%,收率为70.5%。
采用相同的方法,对实施例2、3得到的发酵液进行提取,所得ε-PL及其盐酸盐的纯度分别为97.21%和96.85%,收率分别为71.5%和73.6%。
实施例6:ε-PL的提取
(1)在发酵液储罐中,加入发酵液8倍体积的自来水对实施例1得到的发酵液进行稀释,在室温条件下,利用沉降过滤式离心机对稀释后的发酵液进行离心,获得发酵液离心过滤清液。
(2)将过滤清液经过截留分子量为800KDa的超滤膜,在操作压力0.1MPa条件下浓缩20倍,收集透过液。
(3)将透过液的pH值利用6M NaOH调节至7.0,然后以5BV/h的速率压入装填有大孔弱酸阳离子树脂的一级离子交换柱中进行离子交换;当树脂吸附饱和后,利用去离子水以5BV/h的速率进行洗涤;洗涤结束后,利用1M氨水以5BV/h进行洗脱,收集洗脱液。将洗脱液以5BV/h的速率压入装填有强碱阴离子树脂的二级离子交换柱中进行负吸附,并收集流出液。
(4)利用6M盐酸将流出液pH值调节至8.5左右进行纳滤操作,纳滤膜截留分子量为500Da,操作压力为1.2MPa,并采用一次性添加方式补加去离子水,以除去流出液中大量盐分。待脱盐结束后,再对浓缩液进行2倍浓缩;
(5)将步骤(4)脱盐后的浓缩液加热至90℃,并按质量体积比加入约2%活性炭,在搅拌条件下脱色15min后,经过滤去除活性炭,获得脱色液;
(6)将步骤(5)得到的脱色液进行蒸发浓缩,并将ε-PL浓度浓缩至20%左右;
(7)将步骤(6)得到的浓缩液,进行喷雾干燥,得到ε-PL产品。
按上述提取流程,所得ε-PL及其盐酸盐的纯度为97.10%,收率为75.2%。
采用相同的方法,对实施例2、3得到的发酵液进行提取,所得ε-PL及其盐酸盐的纯度分别为95.13%和96.51%,收率分别为70.2%和74.1%。
对照1:
对实施例1得到的发酵液,采用CN201110053004.2中的方法进行提取,具体是:
在酸化罐中,用2mol/L盐酸调发酵液pH值为5.0,然后加热此酸化液至100℃,保温10分钟,将酸化液冷却至室温,经板框过滤,复滤后得清滤液。加入6mol/L氢氧化钠至清滤液中,调清滤液pH值为7.2得预处理液。
将预处理液泵入大孔强酸型离子交换树脂(D001)吸附柱(高径比为10:1)中进行吸附,吸附过程应控制流速为4mL/min(约4BV/h),直到树脂吸附达饱和状态。用5倍柱体积的去离子水在流速5mL/min(约5BV/h)的条件下洗涤饱和树脂。用预先配制的1mol/L醋酸水溶液进行解析,控制解析速度1mL/min(约1BV/h),在不同保留时间下,接合Itzhaki法或高效液相色谱法收集ε-PL以获得解析液。向含有ε-PL的解析液中加入2.0%(w/w)的活性炭,分别加热升温至100℃,搅拌脱色60分钟,然后冷却至室温,过滤得脱色液。
将ε-PL脱色液压入蒸发浓缩装置进行循环浓缩,浓缩至ε-PL含量在20%(w/w)时停止浓缩。
将浓缩液分别经冷冻干燥可得ε-PL纯品。ε-PL及其盐酸盐的回收率为62.1%,纯度为52.6%。
对照2
省略实施例1中的步骤(2),其他条件与实施例1一致,处理与实施例1相同的发酵液。结果显示,按照此方法所得ε-PL及其盐酸盐的纯度为92.10%,收率为77.2%。
对照3
将实施例1中的步骤(3)进行调整,其他处理条件与实施例1一致,处理与实施例1相同的发酵液。
其中,调整后的步骤(3)具体是:将透过液的pH值利用6M NaOH调节至7.0,然后以5BV/h的速率压入装填有大孔弱酸阳离子树脂的一级离子交换柱中进行离子交换;当树脂吸附饱和后,利用去离子水以5BV/h的速率进行洗涤;洗涤结束后,利用1M氨水以5BV/h进行洗脱,收集洗脱液。
结果显示,按照此方法按上述提取流程,所得ε-PL及其盐酸盐的纯度为93.40%,收率为80.31%。
对照4
省略实施例1中的步骤(4),其他条件与实施例1一致,处理与实施例1相同的发酵液。结果显示,按照此方法所得ε-PL及其盐酸盐的纯度为73.65%,收率为76.86%。
虽然本发明已以较佳实施例公开如上,但其并非用以限定本发明,任何熟悉此技术的人,在不脱离本发明的精神和范围内,都可做各种的改动与修饰,因此本发明的保护范围应该以权利要求书所界定的为准。
- 还没有人留言评论。精彩留言会获得点赞!