一种N-烯丙基丙炔酰胺的合成方法与流程
本发明涉及一种化工、医药和农药的中间体合成方法,具体为一种N-烯丙基丙炔酰胺的合成新方法。
背景技术:
N-烯丙基丙炔酰胺是一种重要的化工、医药和农药中间体,然而由于炔键对胺的敏感性,其合成具有一定的挑战,目前的合成工艺存在原料供应及反应条件苛刻等问题限制着其推广应用。
文献Tetrahedron,2009,65,8733–8737及Tetrahedron Letters,2013,54, 834-839报道了其合成方法,该方法以丙炔酸为原料,在N,N’-二环己基碳二亚胺脱水剂的作用下与烯丙基胺缩合得到N-烯丙基丙炔酰胺,该方法具有明显的缺点:1)需要等摩尔当量甚至更多的缩合剂N,N’-二环己基碳二亚胺,后处理繁琐;2)反应生成大量的副产物1,3-二环己基脲,且难以分离去除干净。
Tetrahedron Letters,1964,23,1477-1480及Synthetic Communications 1993, 23(14), 2003-2010报道了以丙炔酸甲酯为原料通过酯胺交换合成丙炔酰胺的方法。然而,该方法需要-30oC及以下的低温;该方法的最大缺陷是:对于烯丙胺、叔丁胺及苄基胺即使在低温下,也主要生成1,4加成即炔键加成产物。
因此,提供一种反应条件温和易控、反应溶剂体系简单、易处理循环套用、且反应收率高、后处理简单、产品纯度高的N-烯丙基丙炔酰胺的合成新方法,已经是一个值得研究的方法。
技术实现要素:
为了克服上述现有技术中的不足,本发明提供了一种反应条件温和易控、反应溶剂体系简单、易处理循环套用、且反应收率高、后处理简单、产品纯度高的N-烯丙基丙炔酰胺的合成方法。
本发明的目的是这样实现的:
一种N-烯丙基丙炔酰胺的合成方法,由取代的硅基丙炔酸酯和烯丙基胺在溶剂中通过一步反应过程直接制得,反应在发生酯胺交换的同时,伴随脱硅烷化作用,反应式为:
;
所述取代的硅基丙炔酸酯,其中R1包括相同或不同的烷基、卤代烷基、芳基和取代芳基;R2包括烷基、卤代烷基、芳基和取代芳基;
所述的取代的硅基丙炔酸酯和烯丙基胺的投料摩尔比为1︰0.25~4;
所述的取代的硅基丙炔酸酯和烯丙基胺的投料摩尔比优选1︰1;
投料方式为取代的硅基丙炔酸酯单质或溶液滴入烯丙基胺的溶液中;或烯丙基胺单质或溶液滴入取代的硅基丙炔酸酯溶液中;
所述反应溶剂包括卤代烃、水(H2O)、醇、芳烃、醚和极性非质子溶剂中的至少一种;
所述的反应溶剂优选为水;
所述反应溶剂卤代烃包括二氯甲烷、氯仿和1, 2-二氯乙烷等;所述的醇包括甲醇、乙醇、异丙醇和叔丁醇;所述的芳烃包括甲苯、二甲苯和氯苯;所述的醚包括甲醚、乙醚、二乙氧基甲醚和二甲氧基乙醚;所述的极性非质子溶剂包括N, N-二甲基甲酰胺(DMF)、二甲亚砜(DMSO)和四氢呋喃(THF);
所述的制备N-烯丙基丙炔酰胺的反应过程,反应时间为1~20小时,反应温度为-10~40oC,其中优选反应时间为3小时,优选反应温度为0~5oC。
所述的制备N-烯丙基丙炔酰胺的反应过程的后处理方法为:反应结束后反应体系温度调整到室温,反应液用水洗后萃取干燥,负压脱出溶剂,溶剂可循环套用,脱溶后的残留物既为N-烯丙基丙炔酰胺。
积极有益效果:本发明公开的一种化工中间体N-烯丙基丙炔酰胺的新合成方法,以市场易得的取代的硅基丙炔酸酯和烯丙基胺为起始原料,经过一步反应高收率地获得目的产物。该方法通过取代硅基对炔键的保护及脱硅烷化作用,极大地抑制了炔键加成产物的产生,为N-烯丙基丙炔酰胺的获取提供了一种便捷的合成途径。该合成方法所涉及反应过程,反应条件温和易控,反应溶剂体系简单,易处理循环套用。该方法还具有反应收率高、后处理简单、产品纯度高等特点,适于规模生产开发。
附图说明
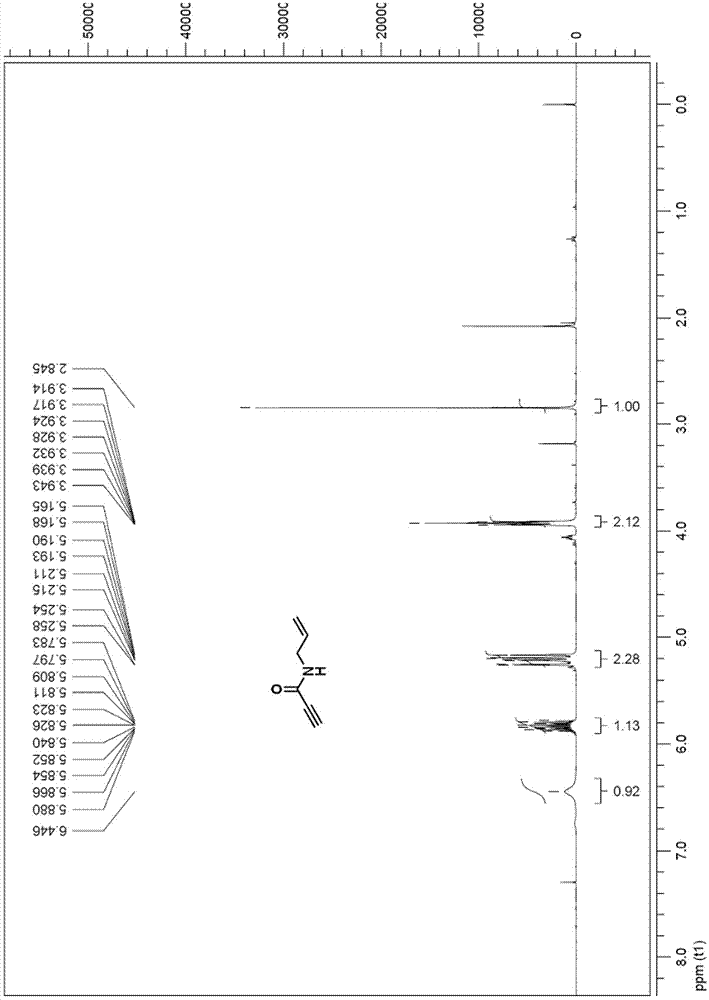
图1为本发明的N-烯丙基丙炔酰胺的1H NMR核磁共振图。
具体实施方式
下面结合具体实施例,对本发明做进一步的说明:
一种N-烯丙基丙炔酰胺的合成方法,由取代的硅基丙炔酸酯和烯丙基胺在溶剂中通过一步反应过程直接制得,反应在发生酯胺交换的同时,伴随脱硅烷化作用,反应式为:
;
所述取代的硅基丙炔酸酯,其中R1包括相同或不同的烷基、卤代烷基、芳基和取代芳基;R2包括烷基、卤代烷基、芳基和取代芳基;
所述的取代的硅基丙炔酸酯和烯丙基胺的投料摩尔比为1︰0.25~4;
所述的取代的硅基丙炔酸酯和烯丙基胺的投料摩尔比优选1︰1;
投料方式为取代的硅基丙炔酸酯单质或溶液滴入烯丙基胺的溶液中;或烯丙基胺单质或溶液滴入取代的硅基丙炔酸酯溶液中;
所述反应溶剂包括卤代烃、水(H2O)、醇、芳烃、醚和极性非质子溶剂中的至少一种;
所述的反应溶剂优选为水;
所述反应溶剂卤代烃包括二氯甲烷、氯仿和1, 2-二氯乙烷等;所述的醇包括甲醇、乙醇、异丙醇和叔丁醇;所述的芳烃包括甲苯、二甲苯和氯苯;所述的醚包括甲醚、乙醚、二乙氧基甲醚和二甲氧基乙醚;所述的极性非质子溶剂包括N, N-二甲基甲酰胺(DMF)、二甲亚砜(DMSO)和四氢呋喃(THF);
所述的制备N-烯丙基丙炔酰胺的反应过程,反应时间为1~20小时,反应温度为-10~40oC,其中优选反应时间为3小时,优选反应温度为0~5oC。
所述的制备N-烯丙基丙炔酰胺的反应过程的后处理方法为:反应结束后反应体系温度调整到室温,反应液用水洗后萃取干燥,负压脱出溶剂,溶剂可循环套用,脱溶后的残留物既为N-烯丙基丙炔酰胺。
实施例1
烯丙基胺(0.57g,10mmol)加入盛有8mL水(H2O)的三口反应瓶中,将温度降到0oC, 然后在0oC下用滴液漏斗缓慢滴加三甲基硅基丙炔酸甲酯(1.56g,10mmol),滴完后,反应体系继续在0oC搅拌反应2小时,气相色谱跟踪,三甲基硅基丙炔酸甲酯转化完全。反应结束后,反应体系温度升到室温。然后加入8mL的二氯甲烷,搅拌、静置、分层。有机相再分别用3毫升水洗涤两次后,用无水硫酸钠干燥后过滤,滤液浓缩后得产品0.93g,含量95%,收率81%。
实施例2
三甲基硅基丙炔酸甲酯(1.56g,10mmol)加入盛有8mL水(H2O)的三口反应瓶中,将温度降到0oC, 然后在0oC下用滴液漏斗缓慢滴加烯丙基胺(0.63g,11mmol),滴完后,反应体系继续在0oC搅拌反应4小时,气相色谱跟踪,三甲基硅基丙炔酸甲酯转化完全。反应结束后,反应体系温度升到室温。然后加入8mL的二氯甲烷,搅拌、静置、分层。有机相再分别用3毫升水洗涤两次后,用无水硫酸钠干燥后过滤,滤液浓缩后得产品0.94g,含量90%,收率78%。
1H NMR (300MHz, CDCl3) δ: 6.4 (br s, 1H);5.87-5.80(m, 1H);5.26-5.17 (dq, J = 12.9, 1.2, 7.5, 0.9, 2H);3.94-3.91 ( m, 2H);2.85(s, 1H)。
本发明以市场易得的取代的硅基丙炔酸酯和烯丙基胺为起始原料,经过一步反应高收率地获得目的产物。该方法通过取代硅基对炔键的保护及脱硅烷化作用,极大地抑制了炔键加成产物的产生,为N-烯丙基丙炔酰胺的获取提供了一种便捷的合成途径。该合成方法所涉及反应过程,反应条件温和易控、反应溶剂体系简单、易处理循环套用。该方法还具有反应收率高、后处理简单、产品纯度高等特点,适于规模生产开发。
以上实施案例仅用于说明本发明的优选实施方式,但本发明并不限于上述实施方式,在所述领域普通技术人员所具备的知识范围内,本发明的精神和原则之内所作的任何修改、等同替代及改进等,均应视为本申请的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!