一种双相酶水解获取宝藿苷I的方法与流程
本发明属于医药技术领域,具体涉及一种双相酶水解获取宝藿苷I的方法。
背景技术:
宝藿苷I(Icariside I,又名淫羊藿次苷II)为小檗科淫羊藿属植物淫羊藿(Epimedium brevicornum Maxim.)中的一种多羟基黄酮类单体成分,有较好的抗肿瘤和抗骨质疏松活性,明显优于淫羊藿中其他黄酮苷类化合物,有被开发成具有良好疗效的抗癌药物的前景。
宝藿苷I是淫羊藿苷脱去一分子葡萄糖的产物(见图1),有报道大鼠口服淫羊藿苷后在粪便和尿液中均检出宝藿苷I,推测大鼠肠菌酶可将淫羊藿苷转化为宝藿苷I,吸收入血的主要为宝藿苷I,亦有文献报道其生物药剂学性质明显优于淫羊藿中其他黄酮类多糖苷,可能是其药效显著的主要原因。
宝藿苷I可以从淫羊藿植物中直接提取,然而淫羊藿中一般以淫羊藿苷形式存在,宝藿苷I的含量很低,只有约0.17 mg/g,且淫羊藿中极性相当的成分很多,分离纯化较困难,产率较低,故单纯提取分离难以满足应用的需求,因此目前主要是通过淫羊藿苷制备宝藿苷I。传统的酸水解或碱水解反应,反应条件苛刻,操作复杂,不利于环境保护,且反应彻底,使得淫羊藿苷及其他黄酮苷类成分上连接的糖全部水解,得到相应的苷元,而不是宝藿苷I等次级苷,所以采用酸碱水解法水解淫羊藿苷或提取物制备宝藿苷I是不可行的,因此目前常采用酶解法。酶水解反应反应条件温和,操作简便,且有利于环境保护,但是酶在有机溶剂中无法发挥效应,而淫羊藿苷在水性酶解体系中溶解度很低,限制了其与酶接触的几率,导致酶解转化周期较长,不利于规模化生产,长期以来相关研究者进行了很多有益的尝试,例如采用超声辅助或者采用环糊精包合技术的方法促进淫羊藿苷溶解,该类技术在一定程度上提高了转化速率和产率,但仍存在以下缺点:一是酶解产物宝藿苷I与淫羊藿苷同时存在,酶解反应无法进行完全;二是操作步骤多,必须采用适当的溶剂把宝藿苷I从水性酶解体系中分离出来;三是酶解反应体系中成分复杂,酶回收困难。
随着宝藿苷I研究的深入,其活性逐渐明确,因此其制备技术也受到越来越多的重视,而适合产业化生产的工艺还未有报道。
技术实现要素:
本发明的目的在于克服现有宝藿苷I制备技术中操作步骤多、成本较高、得率较低、周期长等不足,提供一种基于双相酶水解技术从淫羊藿中直接获取宝藿苷I的方法。
本发明双相酶水解法获取宝藿苷I的方法,实施流程图如图3所示,具体按照下述步骤进行:
(1)称取淫羊藿苷粉,加入醋酸-醋酸钠缓冲溶液或者磷酸盐缓冲溶液,混匀,得到淫羊藿苷混悬液;
(2)步骤(1)所得淫羊藿苷混悬液中加入水解酶,混匀,并加入乙酸乙酯,调节搅拌转速后加热,使得淫羊藿苷在双相溶剂条件下进行水解反应,冷却;
(3)分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I。
其中,步骤(1)中的淫羊藿苷是指淫羊藿苷含量为80%及以上的淫羊藿苷粗品或淫羊藿苷纯品;
所述醋酸-醋酸钠缓冲溶液的pH值介于3.5~5.5;所述磷酸盐缓冲溶液的pH值介于3.5~5.5;
所述淫羊藿苷与缓冲溶液的比例为1 g:100~10000 mL;
步骤(2)中所述的酶为β-葡萄糖苷酶、纤维素酶或β-葡聚糖酶中的一种;
所述淫羊藿苷与酶的体积比为1:40~10:1;
所述淫羊藿苷混悬液与乙酸乙酯的体积比为1:0.5~2.5;
所述搅拌转速为50 rpm~200 rpm,40℃~55℃加热;
所述水解反应时间为0.5 h~48 h。
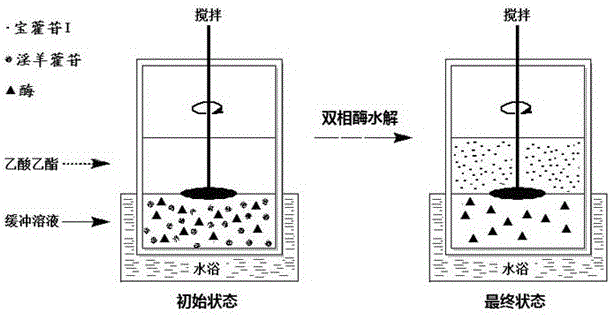
淫羊藿植物中的淫羊藿苷含量较高,在水中溶解度较小,易溶于乙酸乙酯、丙酮等中等极性溶剂,而宝藿苷I在水中几乎不溶,但也易溶于中等极性溶剂。本发明中的双相酶水解法采用β-葡萄糖苷酶、纤维素酶或β-葡聚糖酶水解淫羊藿苷得到宝藿苷I;利用水性酶解体系(水相)和中等极性溶剂(有机相)相互不能混溶的原理构筑双相反应体系;淫羊藿苷大部分存在于有机相,在搅拌条件下不断进入水相发生酶解反应,产物宝藿苷I则快速转移至有机相,促使酶解完全、快速反应,本发明中双相酶水解法的示意图如图2所示。
本发明创新性地将双相体系引入淫羊藿苷制备宝藿苷I中,使有机相成为天然的原料库和产品库,而水相则为生产车间,解决了淫羊藿苷水溶性差导致的酶解慢和不完全的问题。因此本发明中的方法较现有方法更适合于大规模生产宝藿苷I。
本发明的有益效果具体阐述如下:
(1)本发明的制备方法将淫羊藿中淫羊藿苷的水解与宝藿苷I的萃取两步合为一步,简化操作步骤,缩短了制备周期;
(2)本发明的制备方法中双相体系使淫羊藿苷少量且持续进入水相,确保淫羊藿苷与酶较大的接触程度,大大提高了转化速率,减少制备时间;
(3)本发明双相体系中产物宝藿苷I在水相中生成,快速转移至有机相,确保反应完全,同时可减少制备过程中副反应的发生,提高了宝藿苷I得率;
(4)本发明的双相体系使酶用量大大减少,降低了制备成本;
(5)本发明的制备方法可以使淫羊藿苷酶解完全,最终水相和有机相中成分简单,减少了纯化分离过程;
(6)本发明的制备方法采用酶水解淫羊藿苷获取宝藿苷I,制备过程温度在60℃以下,减少了原料和产物分解,且不使用酸碱,有机溶剂可回收再利用,因此对环境几乎没有污染。
附图说明
图1 淫羊藿苷和宝藿苷I分子结构图。
图2 双相酶水解法的示意图。
图3 双相酶水解法制备宝藿苷I工艺的流程图。
图4 酶水解前后高效液相色谱图。
具体实施方式
为使本技术领域人员更好的理解本发明的技术方案,下面结合附图和具体实施例进行描述。
实施例1:
称取淫羊藿苷对照品0.1 g,加入醋酸-醋酸钠缓冲溶液(pH 3.5)200 mL,混匀,得到淫羊藿苷混悬液。上述淫羊藿苷混悬液中加入β-葡萄糖苷酶4.0 g混匀,加入100 mL乙酸乙酯,调节搅拌转速为200 rpm,50℃加热反应0.5 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I产品。酶水解前后高效液相色谱图如图4所示,可见淫羊藿苷在上述条件下已经全部水解为宝藿苷I,得率100%。
实施例2:
称取淫羊藿苷对照品0.1 g,加入醋酸-醋酸钠缓冲溶液(pH 5.5)1000 mL,混匀,得到淫羊藿苷混悬液,上述淫羊藿苷混悬液中加入β-葡萄糖苷酶0.5 g,混匀,加入500 mL乙酸乙酯,调节搅拌转速为50 rpm,40℃加热反应24 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I,得率98.6%。
实施例3:
称取淫羊藿苷对照品0.1 g,加入醋酸-醋酸钠缓冲溶液(pH 5.0)200 mL,混匀,得到淫羊藿苷混悬液,上述淫羊藿苷混悬液中加入β-葡萄糖苷酶2 g,混匀,加入500 mL乙酸乙酯,调节搅拌转速为150 rpm,50℃加热2 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I,得率100%。
实施例4:
称取淫羊藿苷对照品0.1 g,加入磷酸盐缓冲溶液(pH 5.5)200 mL,混匀,得到淫羊藿苷混悬液。上述淫羊藿苷混悬液中加入纤维素酶2.0 g混匀,加入200 mL乙酸乙酯,调节搅拌转速为100 rpm,50℃加热反应3 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I产品,宝藿苷I得率100%。
实施例5:
称取淫羊藿苷提取物细粉1.0 g,加入磷酸盐缓冲溶液(pH 4.0)100 mL,混匀,得到淫羊藿苷混悬液。上述淫羊藿苷混悬液中加入β-葡聚糖酶0.1 g混匀,加入200 mL乙酸乙酯,调节搅拌转速为100 rpm,55℃加热反应24 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I产品,宝藿苷I得率98.0%。
实施例6:
称取淫羊藿苷提取物细粉1.0 g,加入磷酸盐缓冲溶液(pH 5.0)100 mL,混匀,得到淫羊藿苷混悬液。上述淫羊藿苷混悬液中加入β-葡聚糖酶1.0 g混匀,加入250 mL乙酸乙酯,调节搅拌转速为50 rpm,50℃加热反应48 h,冷却,分取乙酸乙酯溶液,减压回收溶剂至干,即得宝藿苷I产品,宝藿苷I得率99.2%。
- 还没有人留言评论。精彩留言会获得点赞!