甲酰胺化合物及其盐形式和用途的制作方法
在先申请的交叉参考本发明要求享有2007年9月12日提交的美国临时申请第60/971,654号;2007年8月2日提交的美国临时申请第60/953,610号;2007年8月2日提交的美国临时申请第60/953,613号;以及2007年8月2日提交的美国临时申请第60/953,614号的权益,通过引用将上述每篇文献全文并入本说明书。
技术领域:
本发明涉及(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺,其新的盐形式,其制备方法,新的中间体,以及治疗广泛多种病况和障碍(包括与中枢神经系统和自主神经系统功能障碍有关的那些)的方法。
背景技术:
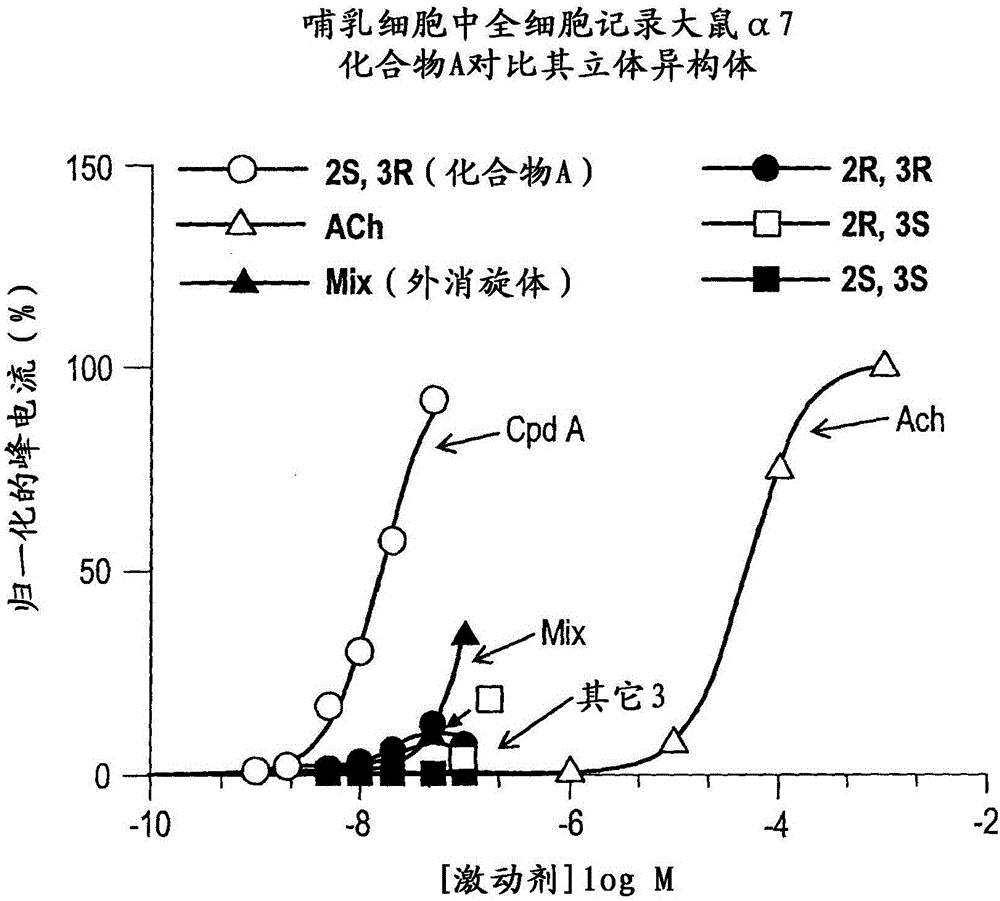
:已表明,中枢神经系统(CNS)特有的神经元烟碱样受体(NNR)以几种亚型存在,其中最常见的亚型是α4β2和α7亚型。例如,参见Schmitt,CurrentMed.Chem.7:749(2000),在此通过引用将其并入本说明书。与α7NNR亚型相互作用的配体已被提议可用于治疗多种病况和障碍。参见Mazurov等人,Curr.Med.Chem.13:1567-1584(2006)和该文献中引用的参考文献,关于α7神经元烟碱样受体亚型的背景理解,通过引用将其并入本说明书。这些病况和障碍中最为突出的有认知损伤、精神分裂症、炎症,血管发生,神经性疼痛和纤维肌痛。在精神分裂症患者的尸体剖检脑组织中,海马NNR数目减少。而且,相对于不吸烟的精神分裂症患者,在吸烟的精神分裂症患者中具有改善的心理效应。烟碱改善动物和精神分裂症患者中的感觉门控缺陷。α7NNR亚型的阻断诱导与精神分裂症中所见的类似的门控缺陷。例如,参见Leonard等人,SchizophreniaBulletin22(3):431(1996),在此通过引用将其并入本说明书。对在具有P50听觉诱发电位门控缺陷的患者中的感觉加工进行的生化、分子和基因研究暗示了α7NNR亚型可能在抑制性神经元途径中起作用。例如,参见Freedman等人,BiologicalPsychiatry38(1):22(1995),通过引用将其并入本说明书。最近,根据Heeschen等人,J.Clin.Invest.100:527(2002)所述,α7NNR被提议是血管发生的介体,通过引用将其并入本说明书。在这些研究中表明,α7亚型的抑制减少了炎性血管发生。而且,α7NNR已被提议作为控制神经发生和瘤生长的靶标(Utsugisawa等人,MolecularBrainResearch106(1-2):88(2002)和美国专利申请2002/0016371,通过引用将它们各自并入本说明书)。最后,最近认识到α7亚型在认知(Levin和Rezvani,CurrentDrugTargets:CNSandNeurologicalDisorders1(4):423(2002)),神经保护(O'Neill等人,CurrentDrugTargets:CNSandNeurologicalDisorders1(4):399(2002)以及Jeyarasasingam等人,Neuroscience109(2):275(2002)),以及神经性疼痛(Xiao等人,Proc.Nat.Acad.Sci(US)99(12):8360(2002))中的作用,通过引用将上述每篇参考文献并入本说明书。据报道,多种化合物与α7NNR相互作用并已根据这一基础被提议作为治疗剂。例如,参见,PCTWO99/62505,PCTWO99/03859,PCTWO97/30998,PCTWO01/36417,PCTWO02/15662,PCTWO02/16355,PCTWO02/16356,PCTWO02/16357,PCTWO02/16358,PCTWO02/17358,Stevens等人,Psychopharm.136:320(1998),Dolle等人,J.LabelledComp.Radiopharm.44:785(2001)和Macor等人,Bioorg.Med.Chem.Lett.11:319(2001)和其中所引用的参考文献,关于α7NNR和被提议的治疗剂的背景教导,通过引用将上述参考文献并入本说明书。在这些化合物中,共同的结构主题是被取代的叔双环胺的结构(例如奎宁环)。也报道了类似的被取代的奎宁环化合物与毒蕈碱性受体结合。例如,参见Sabb的美国专利5,712,270以及PCTWO02/00652和PCTWO02/051841,关于这些化合物,通过引用将每篇参考文献并入本说明书。一些烟碱样化合物的限制是它们伴有各种不希望的副作用,例如通过刺激肌肉和神经节受体而引起的那些副作用。继续需要用于预防或治疗各种病况或障碍诸如CNS障碍,包括缓解这些障碍的症状的化合物、组合物和方法,其中所述化合物表现出具有有益效果的烟碱样药理学,即,影响CNS发挥作用,但无显著的有关副作用。仍然需要影响CNS功能而不显著影响那些可能诱导不希望的副作用诸如在心血管和骨骼肌部位处的明显活性的烟碱样受体亚型的化合物、组合物和方法。本发明提供了这类化合物、组合物和方法。发明概述本发明的一个方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。本发明的另一个方面是实质上纯形式的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。本发明的另一个方面是实质上不含(2S,3S)、(2R,3S)或(2R,3R)异构体的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。另外,本发明的另一个方面是立体异构体富集的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。在一个实施方案中,对映体和/或非对映体过量是90%或更大。在一个实施方案中,对映体和/或非对映体过量是95%或更大。在一个实施方案中,对映体和/或非对映体过量是98%或更大。在一个实施方案中,对映体和/或非对映体过量是99%或更大。在一个实施方案中,对映体和/或非对映体过量是99.5%或更大。本发明的另一个方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的酸盐,其中所述酸选自:盐酸,硫酸,磷酸,马来酸,对甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟脑磺酸,酮戊二酸或马尿酸。在一个实施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺与酸的化学计量是2:1、1:1或1:2。在一个实施方案中,所述化学计量是1:1。本发明的一个实施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺盐酸盐或其水合物或溶剂合物,包括部分水合物或部分溶剂合物。另一个实施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐或其水合物或溶剂合物,包括部分水合物或部分溶剂合物。本发明还提供了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺和新的中间体的可放大规模的合成。本发明的范围包括本说明书所述的各方面、实施方案和优先选择的所有组合。附图说明图1A1-1A4图示了在哺乳动物GH4C1细胞中表达的大鼠α7受体对(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺;外消旋物,即(2S,3R)、(2R,3S)、(2R,3R)和(2S,3S)的混合物;单独的立体异构体;以及乙酰胆碱(ACh)的应答。图1B图示了在哺乳动物GH4C1细胞中表达的大鼠α7受体对处于有效血浆浓度范围内的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺;外消旋物,即(2S,3R)、(2R,3S)、(2R,3R)和(2S,3S)的混合物;以及单独的立体异构体的功能应答的比较。图2A图示了在非洲蟾蜍卵母细胞中表达的人α7受体对(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的应答。图2B图示了在施用所示浓度的化合物后人α7受体的对照应答。将数据归一化为在实验激动剂诱发的应答前5分钟所获得的对照300μMACh应答的净电荷。每个点代表至少4个卵母细胞的归一化应答的平均数±SEM。图3图示了在物体识别(OR)模型中的认知作用的评价,证明了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺以0.3和1mg/kg腹膜内给药时具有积极影响,*p<0.5。图4图示了在OR模型中的认知作用的评价,证明了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在宽的剂量范围(0.3-10mg/kg)内口服给药时具有积极影响,*p<0.5。图5图示了腹膜内给药的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在OR任务中在预防由MK-801(亦称地佐环平,一种市售的NMDA受体的非竞争性拮抗剂)诱导的认知缺陷中的作用。图6图示了在OR任务中由用介质处理的组在最后的亚急性给药(经口给药)后30分钟、6小时或24小时在物体A上所花费的平均时间相对于在物体B上所花费的平均时间没有显著差别(分别是p=0.17,p=0.35和p=0.12)。或者,在最后的亚急性给药0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺后30分钟、2小时、6小时和18小时,受试者探索物体B(新的物体)比探索物体A(熟悉的物体)花费显著(P<0.05)更多的时间。另外,在用0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺处理的动物中的认知指数在2小时(75%)和6小时(71%)显著高于在最后给药后30分钟时用介质处理的组的认知指数(54%)。图7图示了在放射状臂形迷宫(RAM)模型中的认知作用的评价。在每日训练期间之前30分钟口服给药(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.1、0.3和1.0mg/kg)。在给药的第二周期间,在用0.3mg/kg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺处理的组中,任务的行为表现的改善是明显的。图8图示了作为由多巴胺过度刺激所诱导的活动过强行为被测量的抗精神病效果的研究,表明(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.3和1.0mg/kg;皮下给药)在大鼠中皮下给药后减弱由阿扑吗啡(1.0mg/kg)所诱导的运动活性过强。图9图示了抗精神病评价,前脉冲抑制,显示由阿扑吗啡诱导的缺陷在皮下给药后被(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的预处理所逆转。图10A图示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐的X射线结晶学分析的结果,确认了该物质的绝对立体化学。被描绘的化合物是部分水合的盐酸盐,完全有序的氯离子和在不对称单元中部分占据的水分子显示了这一点。图10B图示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐的X射线结晶学分析的结果,确认了该物质的绝对立体化学,使用编号方案描述供参考。视图是向下观察晶胞的结晶学b-轴。分子之间的氢键用虚线表示。图11A图示了(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺对氯苯甲酸盐的X射线结晶学分析的结果,确认了该物质的绝对立体化学。图11B图示了(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺对氯苯甲酸盐的X射线结晶学分析的结果,确立了该物质的绝对立体化学,使用编号方案描述供参考。图12图示了表征N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的四种立体异构体的完整色谱图,其中2S,3R在5.3分钟保留时间处显示出峰,2R,3S在7.3分钟的保留时间处显示出峰,2R,3R在8.2分钟的保留时间处显示出峰,以及2S,3S在12.4分钟的保留时间处显示出峰。如本说明书中所述,分析以提供了足够的分辨率所需的流动相形成60:40:0.2的己烷:乙醇:二正丁胺的组合物,流速为1.0毫升/分钟,柱温为20℃,以及UV检测波长是270nm。图13是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐的XRPD,图示了观察图形(较亮)和计算图形(较暗)。两个图形在2θ值方面是一致的,在强度和峰宽方面的较小差别可归于仪器分辨率和优选取向效应。如本说明书中所述,其它较小差异可归因于由于在室温下被收集的观察数据和从120K的结构计算的数据所导致的温度变动。图14是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一甲苯磺酸盐的XRPD。发明的详细说明对α7NNR亚型具有亲合力(≤1nMKi值)和选择性的具体化合物(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在认知(认知增强)和精神病(抗精神病效果)的动物模型中显示了功效。本发明的一个方面是(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。另一个方面是实质上纯形式的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。另一个方面是实质上不含(2S,3S)、(2R,3S)或(2R,3R)异构体的(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。另外,另一个方面是立体异构体富集的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐。在一个实施方案中,对映体和/或非对映体过量是90%或更大。在一个实施方案中,对映体和/或非对映体过量是95%或更大。在一个实施方案中,对映体和/或非对映体过量是98%或更大。在一个实施方案中,对映体和/或非对映体过量99%或更大。在一个实施方案中,对映体和/或非对映体过量是99.5%或更大。本发明的另一个方面是(2S,3R)N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的酸盐,其中所述酸选自:盐酸,硫酸,磷酸,马来酸,对甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟脑磺酸,酮戊二酸或马尿酸。在一个实施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺与酸的化学计量是2:1、1:1或1:2。在一个实施方案中,所述化学计量是1:1。本发明的一个实施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺盐酸盐或其水合物或溶剂合物,包括部分水合物或部分溶剂合物。另一个实施方案是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐或其水合物或溶剂合物,包括部分水合物或部分溶剂合物。本发明的另一个方面是(2S,3R)-(2-((3-吡啶基)甲基)-3-氨基-1-氮杂双环[2.2.2]辛烷。本发明的另一个方面是包含本发明的化合物和一种或多种药学上可接受的载体的药物组合物。本发明的另一个方面是治疗或预防中枢神经系统障碍、炎症、疼痛或新血管形成方法,它包括给予本发明的化合物。在一个实施方案中,所述中枢神经系统障碍以正常神经递质释放的改变为特征。在一个实施方案中,所述中枢神经系统障碍选自轻度认知损伤、年龄相关的记忆损伤、早老性痴呆、早发性阿尔茨海默病、老年性痴呆、阿尔茨海默病型痴呆、阿尔茨海默病、莱维体(LewyBody)痴呆、微梗塞性痴呆、AIDS相关性痴呆、HIV痴呆、多发性脑梗塞、帕金森神经功能障碍、帕金森病、皮克病、进行性核上性麻痹、亨廷顿舞蹈病、迟发性运动障碍、运动机能亢进、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、抑郁症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。在一个实施方案中,所述中枢神经系统障碍选自阿尔茨海默病、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。本发明的另一个方面包括本发明的化合物在制备用于治疗或预防中枢神经系统障碍、炎症、疼痛或新血管形成的药物中的用途。在一个实施方案中,所述中枢神经系统障碍以正常神经递质释放的改变为特征。在一个实施方案中,所述中枢神经系统障碍选自轻度认知损伤、年龄相关的记忆损伤、早老性痴呆、早发性阿尔茨海默病、老年性痴呆、阿尔茨海默病型痴呆、阿尔茨海默病、莱维小体痴呆、微梗塞性痴呆、AIDS相关性痴呆、HIV痴呆、多发性脑梗塞、帕金森神经功能障碍、帕金森病、皮克病、进行性核上性麻痹、亨廷顿舞蹈病、迟发性运动障碍、运动机能亢进、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、抑郁症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。在一个实施方案中,所述中枢神经系统障碍选自阿尔茨海默病、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。本发明的另一个方面是用于治疗或预防中枢神经系统障碍、炎症、疼痛或新血管形成的本发明的化合物。在一个实施方案中,所述中枢神经系统障碍以正常神经递质释放的改变为特征。在一个实施方案中,所述中枢神经系统障碍选自轻度认知损伤、年龄相关的记忆损伤、早老性痴呆、早发性阿尔茨海默病、老年性痴呆、阿尔茨海默病型痴呆、阿尔茨海默病、莱维小体痴呆、微梗塞性痴呆、AIDS相关性痴呆、HIV痴呆、多发性脑梗塞、帕金森神经功能障碍、帕金森病、皮克病、进行性核上性麻痹、亨廷顿舞蹈病、迟发性运动障碍、运动机能亢进、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、抑郁症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。在一个实施方案中,所述中枢神经系统障碍选自阿尔茨海默病、躁狂症、注意缺陷障碍、注意涣散多动症、焦虑症、诵读困难、精神分裂症、精神分裂症中的认知功能障碍、抑郁症、强迫观念与行为障碍或图雷特综合征。在上述的方法和用途中,在本发明的一个实施方案中,有效剂量为每24小时期间约1毫克至10毫克。本发明的另一个方面是通过(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-酮的依次动力学拆分和立体选择性还原胺化,制备(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐的方法。本发明的范围包括本说明书所述的各方面、实施方案和优先选择的所有组合。药物候选物的商业性开发涉及许多步骤,包括按比例放大化学合成和纯化,发现最佳的盐形式,等等。在药物组合物的制剂中,药物物质优选处于可方便地进行处理和加工的形式。需要考虑的事项包括商业可行性以及制造的一致性。另外,在制备药物组合物中,重要的是在给药至患者后,提供可靠的、可重现的和恒定的药物血浆浓度曲线。活性成分的化学稳定性、固态稳定性和“储存期限”也是非常重要的因素。这些药物物质和包含这些药物物质的组合物应优选能够在可估计的时段内被有效保存,在活性组分的物化特征方面(例如,其化学组成,密度、吸湿性和溶解性)不表现出重大变化。另外,同样重要的是能够提供尽可能化学纯的形式的药物。在下文中更详细地讨论本发明的这些特征。I.化合物本发明的化合物是由以下化合物A表示的(2S,3R)-N(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或化合物A的药学上可接受的盐的形式。在Mazurov等人的美国专利No.6,953,855中描述了外消旋化合物N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺、合成和医学治疗的效用,在此通过引用将其并入本说明书。外消旋的N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺是神经元烟碱样受体(NNR)的α7亚型的高亲和力配体。外消旋的N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺含有两个不对称取代的碳原子。因此,外消旋的N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺以四种立体异构体形式即(S,S)、(S,R)、(R,R)和(R,S)存在。(S,R),即(2S,3R),是化合物A。先前认为,在报道的合成(包括美国专利6,953,855)中产生的主要的立体异构形式以在1-氮杂双环[2.2.2]辛烷(奎宁环)环的2和3位处的顺式相对构型为特征。换句话说,认为顺式非对映体(对映体的(2R,3R)和(2S,3S)对)是根据报道的方法制备时得到的主要形式。这一主要顺式合成的确定基于:(i)奎宁环以及分离的非对映(顺式和反式)中间体的2位和3位氢核的1H偶合常数与文献报道的偶合常数的比较;以及(ii)通过类似于文献所述,所述文献如Warawa等人,J.Med.Chem.18(6):587-593(1975)和Viti等人,LetrahedronLett.35(32):5939-5942(1994),两篇文献都通过引用将其并入本说明书,用于产生化合物混合物的合成化学的预期立体化学结果。因此,预料到将形成顺式构型。就这点而论,使用外消旋物进行生物试验所产生的结果被假定为归因于主要的顺式构型。目前借助于结晶盐形式和类似物的X射线衍射分析已经发现,在最初合成中产生的主要的非对映体实际上是反式非对映体。另外,已经发现具有反式相对立体化学的两种对映体即(2S,3R)和(2R,3S),在它们与α7NNR亚型相互作用的能力方面彼此实质上不同。(2S,3R)构型,即化合物A,具有更大的活性。通过进一步的分析,已经发现化合物A具有使其与:i)单独获得的其它三种立体异构体中的每一种,ii)所有四种立体异构体的混合物,即外消旋物;以及iii)在文献中报道的其它α7NNR配体相区别的药理学性质。(2S,3R)-N(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(化合物A)是在α7NNR受体处的高度选择性的完全激动剂,其具有显著低的EC50(用于激活)值并且在EC50和IC50(用于残余抑制)之间具有良好的距离差,在宽的治疗可用浓度范围内提供了功能激动。II.(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的可放大规模合成具体的合成步骤在它们按比例放大的可行性方面有差异。发现由于多种原因而使得反应缺乏按比例放大的能力,所述原因包括安全考虑、试剂费用、困难的后处理或纯化、反应能学(热力学或动力学)和反应收率。本说明书所述的(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的合成已用于生成千克量级的材料,并且已经以高收率在数千克规模上进行了组分反应。可按比例放大的合成采用了可外消旋的酮(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮)的动态拆分和经过拆分的酮的(R)-α-甲基苄基胺亚胺衍生物的立体选择性还原(还原胺化)。本说明书所报道的合成顺序可容易地进行规模放大并免除色谱纯化。III.(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的新的盐形式的制备(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺作为游离碱是具有非常有限的水溶性的无定形粉末。该游离碱与无机酸和有机酸反应以生成某些酸加成盐,这些酸加成盐具有对于药物组合物的制备而言是有利的物理和化学性质,包括但不限于结晶度、水溶性和稳定性。本发明的盐的化学计量可以改变。根据形成本说明书所述盐的方式,盐可具有吸留了在成盐期间所存在的溶剂的晶体结构。因此,盐可以作为相对于(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺具有不同化学计量的溶剂的水合物和其它溶剂合物存在。制备盐形式的方法可以改变。(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺盐形式的制备一般包括:(i)将游离碱或游离碱的溶液即(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在合适的溶剂中的溶液与纯酸混合,或者与该酸在合适的溶剂中的溶液混合;(iia)如有必要,将得到的盐溶液冷却,以引起沉淀;或(iib)加入合适的抗溶剂以引起沉淀;或(iic)蒸发第一溶剂并加入新的溶剂并重复步骤(iia)或步骤(iib);以及(iii)过滤和收集得到的盐。在一个实施方案中,(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺是立体异构体富集的。在一个实施方案中,对映体和/或非对映体过量是90%或更大。在一个实施方案中,对映体和/或非对映体过量是95%或更大。在一个实施方案中,对映体和/或非对映体过量是98%或更大。在一个实施方案中,对映体和/或非对映体过量是99%或更大。在一个实施方案中,对映体和/或非对映体过量是99.5%或更大。使用的化学计量、溶剂配合比、溶质浓度和温度可以改变。可用于盐形式的制备或重结晶的代表性溶剂包括但不限于乙醇、甲醇、异丙醇、丙酮、乙酸乙酯和乙腈。合适的药学上可接受的盐的实例包括无机酸加成盐诸如氯化物,溴化物,硫酸盐,磷酸盐和硝酸盐;有机酸加成盐诸如乙酸盐,半乳糖二酸盐,丙酸盐,琥珀酸盐,乳酸盐,羟乙酸盐,苹果酸盐,酒石酸盐,柠檬酸盐,马来酸盐,富马酸盐,甲磺酸盐,对甲苯磺酸盐和抗坏血酸盐;以及与氨基酸形成的盐诸如天冬氨酸盐和谷氨酸盐。盐有时候可为水合物或乙醇溶剂合物。代表性的盐根据Dull等人的美国专利第5,597,919号,Dull等人的美国专利第5,616,716号和Ruecroft等人的美国专利第5,663,356号所述被提供,通过引用将它们各自并入本说明书。对于游离碱(2S,3R)-N-(2-((3-吡啶基)甲基-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺进行的盐筛选显示,尽管可形成许多的药学上可接受的酸的盐,但是这些盐中只有少数具有对于商业制造而言是可接受的性质。因此,不存在预测由商业可行的盐所体现的特征的能力。提供作为结晶的盐(即显示一定的结晶度的盐)的酸,根据制备盐方法,包括盐酸,硫酸,磷酸,对甲苯磺酸,半乳糖二酸(粘酸),D-扁桃酸,D-酒石酸,甲磺酸,R-和S-10-樟脑磺酸,马来酸,酮戊二酸和马尿酸。在这些盐中,盐酸、磷酸、马来酸和对甲苯磺酸的盐各自表现出另外的合乎需要的性质,包括高熔点、良好的水溶性和低吸湿性。这些盐中的这些特征是意想不到的。IV.药物组合物本发明的药物组合物包括本说明书所述的盐,所述盐为纯态或为其中所述化合物与任何其它药学相容的产品(其可为惰性的或具生理学活性的)相组合的组合物形式。得到的药物组合物可用于预防易患病况或障碍的受试者中的所述病况或障碍,以及/或治疗罹患所述病况或障碍的受试者。本说明书的药物组合物包括本发明的化合物和/或其药学上可接受的盐。化合物的给药方式可以改变。所述组合物优选口服给药(例如,在溶剂诸如水性或非水性液体中的液体形式中,或在固体载体中)。用于口服给药的优选组合物包括丸剂,片剂,胶囊,胶囊性片剂,糖浆剂和溶液,包括硬明胶胶囊和时间释放胶囊。标准的赋形剂包括粘合剂、填充剂、着色剂、增溶剂等等。组合物可被配制成单位剂量形式,或为多剂量或亚单位剂量。优选的组合物为液体或半固体形式。可以使用包括液体药学惰性载体诸如水或其它药学相容的液体或半固体的组合物。这些液体和半固体的使用是本领域技术人员公知的。组合物还可通过注射即静脉内、肌内、皮下、腹膜内、动脉内、鞘内;以及脑室内途径给药。静脉内给药是优选的注射方法。合适的注射用载体是本领域技术人员公知的并且包括5%的葡萄糖溶液、盐水和磷酸盐缓冲盐水。药物产品还可作为输注剂或注射剂给予(例如作为在药学上可接受的液体或液体混合物中的悬浮液或乳液)。制剂还可使用其它手段例如直肠给药进行给药。可用于直肠给药的制剂诸如栓剂是本领域技术人员公知的。药物产品还可通过吸入(例如,以气雾剂形式经鼻或使用Brooks等人的美国专利第4,922,901号中所述类型的递送制品进行,将该文献全文并入本说明书);局部(例如,以洗剂形式);透皮(例如,使用透皮贴剂)或离子导入方式给药;或通过舌下或经颊给药。尽管有可能给予呈本体活性化学品形式的化合物,但是优选提供用于有效率的和有功效的给药的药物组合物或药物制剂形式的药物产品。化合物给药的示例性方法是本领域技术人员公知的。这些制剂的有用性可根据所用的具体组合物和接受治疗的具体受试者的不同而异。这些制剂可含有液体载体,该液体载体可以是油性的、水性的、被乳化的或含有某些适合于给药方式的溶剂。组合物可以周期性地或以渐变的、连续的、恒定的或受控的速率被给予至温血动物(例如哺乳动物,诸如小鼠,大鼠,猫,兔,狗,猪,牛或猴),但是有利地被给予至人类。另外,药物制剂的给药天数和每天给药次数可以改变。用于本发明化合物给药的其它合适的方法描述于Smith等人美国专利第5,604,231号中,通过引用将该文献的内容并入本说明书。在本发明的实施方案中并且可由本领域技术人员所理解的,本发明的化合物可与其它的治疗化合物组合给药。例如,本发明的化合物可与诸如以下的物质组合使用:其他的NNR配体(诸如伐尼克兰),抗氧化剂(诸如自由基清除剂),抗细菌药(诸如青霉素类抗生素),抗病毒药(诸如核苷类似物,如齐多夫定和阿昔洛韦),抗凝血药(诸如华法林),抗炎药(诸如NSAID),解热药,镇痛药,麻醉药(诸如手术中使用的麻醉药),乙酰胆碱酯酶抑制剂(诸如多奈哌齐和加兰他敏),抗精神病药(诸如氟哌啶醇,氯氮平,奥氮平和喹硫平),免疫抑制剂(诸如环孢菌素和甲氨蝶呤),神经保护药,甾族化合物(诸如甾体激素),皮质类固醇(诸如地塞米松,强的松和氢化可的松),维生素,矿物质,营养制品,抗抑郁药(诸如丙米嗪,氟西汀,帕罗西汀,依他普仑,舍曲林,文拉法辛和度洛西汀),抗焦虑药(诸如阿普唑仑和丁螺环酮),抗惊厥药(诸如苯妥英和加巴喷丁),血管扩张药(诸如哌唑嗪和西地那非),情绪稳定药(诸如丙戊酸盐和阿立哌唑),抗癌药(诸如抗增殖药),抗高血压药(诸如阿替洛尔,可乐定,氨氯地平,维拉帕米和奥美沙坦),轻泻药,粪便软化药,利尿药(诸如呋塞米),解痉药(诸如双环维林),抗运动障碍药和抗溃疡药(诸如艾美拉唑)。本发明的化合物可单独使用或与其它的治疗剂组合使用。药学活性剂的这种组合可以一起或分开进行给药,并且当分开给药时,给药可以同时进行或以任何顺序依次进行。化合物或药剂的量以及给药的相对时机将经过选择,以便获得所需的治疗效果。组合给药可以是以下形式的同时给药:(1)包括多种化合物的整体式药物组合物;或(2)单独的药物组合物,其各自包含所述化合物中的一种。或者,所述组合可以顺序方式分开给药,在该顺序方式中,先给予一种治疗剂,然后给予另一种治疗剂,或者反之亦然。该顺序给药在时间上可以间隔较近或较远。本发明的化合物可用于治疗多种障碍和病况,并且因此,本发明的化合物可与多种其它合适的可用于治疗或预防这些障碍或病况的治疗剂组合使用。化合物的合适的剂量是有效防止罹患障碍的患者发生所述障碍的症状或治疗所述障碍的一些症状的量。所提到的“有效量”、“治疗量”或“有效剂量”是指足以引发所需的药理学或治疗学效果从而导致有效预防或治疗障碍的量。当治疗CNS障碍时,化合物的有效量是足以穿过受试者的血脑屏障以便结合至该受试者的脑中的有关受体部位并调节有关NNR亚型的活性(例如提供神经递质分泌,从而导致有效预防或治疗障碍)的量。障碍的预防的实例体现为延迟障碍的症状的发生。障碍的治疗的实例体现为减少与障碍有关的症状或改善障碍症状的复发。优选地,有效量足以获得所需的结果,但不足以引起明显的副作用。有效剂量可根据诸如患者的状况、障碍的症状的严重程度和药物组合物的给药方式的因素不同而异。对于人类患者,典型化合物的有效剂量一般要求给予一定量的化合物,该量足以调节有关的NNR的活性,但是该量应不足以诱导对骨骼肌和神经节的影响达到任何显著程度。化合物的有效剂量当然因患者与患者之间的不同而异,但是通常包括从以发生CNS效果或其它所需治疗效果为起点、但低于观察到肌肉效果的量的范围内的量。本说明书所述的化合物,当根据本说明书所述方法以有效量被使用时,可以提供一定程度的对CNS障碍或其它障碍的进展的预防、CNS障碍或其它障碍的症状的改善,或使CNS障碍或其它障碍的复发改善达到一定程度。这些化合物的有效量通常低于引发任何明显的副作用(例如,与骨骼肌或神经节有关的那些副作用)所需的阈值浓度。化合物可在某些CNS障碍和其它障碍被治疗并且某些副作用可被避免的治疗窗内进行给药。理想地是,本说明书所述化合物的有效剂量足以为障碍提供所希望的效果但是不足以(即,不在足够高的水平下)提供不希望的副作用。优选地,化合物在有效治疗CNS障碍或其它障碍但是低于引发某些副作用到任何显著程度所需的量的1/5、通常低于1/10的剂量下被给药。最优选地,有效剂量为当观察到发生最大效果并具有最小副作用时的极低浓度。典型地,这些化合物的有效剂量一般要求以低于5mg/kg患者重量的量给予化合物。通常,本发明的化合物以低于约1mg/kg患者体重被给予,通常以低于约100μg/kg患者重量被给予,但是经常以约10μg到低于100μg/kg患者重量被给予。上述的有效剂量一般地代表作为单剂量被给予的量,或作为在24小时期间以一个或多个剂量被给予的量。对于人类患者,典型化合物的有效剂量一般要求以至少约1毫克/24小时/患者、通常至少约10毫克/24小时/患者、并且经常至少约100毫克/24小时/患者的量给予化合物。对于人类患者,典型化合物的有效剂量要求给予一般不超过约500毫克/24小时/患者、通常不超过约400毫克/24小时/患者、并且经常不超过约300毫克/24小时/患者的量的化合物。另外,组合物可有利地以有效剂量被给予,从而使得化合物在患者血浆内的浓度通常不超过50ng/mL,通常不超过30ng/mL,并且经常不超过10ng/mL。在本发明的一个实施方案中,在24小时期间的有效剂量为约1mg到10mg。IV.药物组合物的用法本说明书使用的“激动剂”是刺激其结合配对物(通常是受体)的物质。刺激在具体试验的背景下进行定义,或者根据文献讨论而是清楚明白的,其在本领域技术人员可理解的实质上类似的环境下进行了被接受为具体结合配对物的“激动剂”或“拮抗剂”的因子或物质的比较。刺激可根据由激动剂或部分激动剂与结合配对物的相互作用所诱导的具体效果或功能的增加来定义并且可包括变构作用。本说明书使用的“拮抗剂”是抑制其结合配对物(通常是受体)的物质。抑制在具体试验的背景下进行定义,或者根据文献讨论而是清楚明白的,其在本领域技术人员可理解的实质上类似的环境下进行了被接受为具体结合配对物的“激动剂”或“拮抗剂”的因子或物质的比较。抑制可根据由拮抗剂与结合配对物的相互作用所诱导的具体效果或功能的降低来定义并且可包括变构作用。本说明书使用的“部分激动剂”或“部分拮抗剂”分别是为其结合配对物分别提供非全部的或不完全的激动或拮抗效果的刺激或抑制水平。可认识到,抑制以及由此的抑制被固有地定义用于要被定义为激动剂、拮抗剂或部分激动剂的任何物质或任何类别的物质。本说明书使用的“固有活性”或“功效”涉及结合配对物复合物的生物有效性的一些量度。关于受体药理学,固有活性或功效被定义的背景应当根据结合配对物(例如受体/配体)复合物的背景和与特定的生物结果有关的活性的考虑而定。例如,在一些情况下,固有活性可根据所涉及的特定的第二信使系统的不同而异。参见Hoyer,D.和Boddeke,H.,TrendsPharmacol.Sci14(7):270-5(1993),关于这一教导,通过引用将其并入本说明书。这些前后关联的具体评价在什么情况下相关联以及它们在本发明的背景下如何相关联,是本领域技术人员显而易见的。本说明书使用的受体调节包括受体的激动,部分激动,拮抗,部分拮抗或反向激动。本说明书使用的其释放受到本说明书所述化合物介导的神经递质包括但不限于乙酰胆碱,多巴胺,去甲肾上腺素,5-羟色胺和谷氨酸,并且本说明书所述的化合物起到CNSNNR的α7亚型的调节剂的作用。本说明书使用的术语“预防”或“防止”包括疾病、障碍或病况进展的任何程度的减少,或疾病、障碍或病况发病的任何程度的延迟。该术语包括提供了对抗具体的疾病、障碍或病况的保护效果以及包括疾病、障碍或病况的复发的改善。因此,在另一个方面,本发明提供了治疗患有NNR或nAChR介导的障碍的患者或者有发展或经历由NNR或nAChR介导的障碍的复发危险的受试者的方法。本发明的化合物和药物组合物可用于实现有益的治疗或预防效果,例如,在罹患CNS功能障碍的受试者中。如上所述,本发明的游离碱和盐化合物调节CNS所特有的α7NNR亚型,并且可通过调节α7NNR用于预防或治疗罹患或易患多种病况或障碍的受试者中的该病况或障碍,包括CNS病况或障碍。化合物能够选择性结合于α7NNR并表达烟碱性药理学,例如,如所述的那样充当激动剂、部分激动剂、拮抗剂。例如,本发明的化合物当以有效量被给予至有需要的患者时,提供了CNS障碍进展的一定程度的预防,即,提供了保护作用,改善了CNS障碍的症状,或改善了CNS障碍的复发,或其组合。本发明的化合物可用于治疗或预防其它类型的烟碱化合物已被提议或显示可用作治疗剂的那些类型的病况和障碍。例如,参见上文所列举的文献,以及Williams等人,DrugNewsPerspec.7(4):205(1994),Arneric等人,CNSDrugRev.1(1):1-26(1995),Arneric等人,Exp.Opin.Invest.Drugs5(1):79-100(1996),Bencherif等人,J.Pharmacol.Exp.Ther.279:1413(1996),Lippiello等人,J.Pharmacol.Exp.Ther.279:1422(1996),Damaj等人,J.Pharmacol.Exp.Ther.291:390(1999);Chiari等人,Anesthesiology91:1447(1999),Lavand'hommeandEisenbach,Anesthesiology91:1455(1999),Holladay等人,J.Med.Chem.40(28):4169-94(1997),Bannon等人,Science279:77(1998),PCTWO94/08992,PCTWO96/31475,PCTWO96/40682,以及Bencherif等人的美国专利第5,583,140号,Dull等人的美国专利第5,597,919号,Smith等人的美国专利第5,604,231号和Cosford等人的美国专利第5,852,041号,关于这些治疗教导,通过引用将它们的内容并入本说明书。化合物及其药物组合物可用于治疗或预防多种CNS障碍,包括神经变性障碍、神经精神病学障碍、神经学障碍和成瘾。化合物及其药物组合物可用于治疗或预防年龄相关性及其它认知缺陷和功能障碍;注意性障碍和痴呆,包括由感染因子或代谢紊乱所引起的那些;提供神经保护;治疗惊厥和多发性脑梗塞;治疗心境障碍,强迫症和成瘾行为;提供痛觉丧失;控制炎症,诸如由细胞因子和核因子κB所介导的炎症;治疗炎性障碍;提供疼痛缓解;治疗代谢障碍诸如糖尿病或代谢综合征;治疗感染,作为用于治疗细菌、真菌和病毒感染的抗感染药。CNS障碍可使用本发明的化合物和药物组合物进行治疗或预防的障碍、疾病和病况有:年龄相关性记忆损伤(AAMI)、轻度认知损伤(MCI)、年龄相关性认知下降(ARCD)、早老性痴呆、早发性阿尔茨海默病、老年性痴呆、阿尔茨海默病型痴呆、阿尔茨海默病、无痴呆的认知损伤(CIND)、莱维小体痴呆、HIV痴呆、AIDS痴呆综合征、血管性痴呆、唐氏(Down)综合征、头创伤、创伤性脑损伤(TBI)、拳击运动员痴呆、克-雅(Creutzfeld-Jacob)病和朊病毒疾病、卒中、缺血、注意缺陷障碍、注意涣散多动症、诵读困难、精神分裂症、精神分裂症样障碍、分裂情感性障碍、精神分裂症中的认知功能障碍、精神分裂症中的认知缺陷诸如记忆(包括工作记忆)、执行功能、注意力、警惕性、信息处理和学习、与精神分裂症有关的痴呆(无论是轻度、中度还是重度痴呆)、与精神分裂症有关的痴呆(无论是轻度、中度还是重度痴呆)、帕金森神经功能障碍、包括帕金森病、脑炎后帕金森神经功能障碍、关岛(Gaum)帕金森神经功能障碍-痴呆、帕金森病型额颞叶痴呆(FTDP)、皮克(Pick)病、尼-皮(Niemann-Pick)病、亨廷顿病、亨廷顿舞蹈病、迟发性运动障碍、运动机能亢进、进行性核上性麻痹、进行性核上性局部麻痹、不宁腿综合征、多发性硬化、肌萎缩性侧索硬化(ALS)、运动神经元疾病(MND)、多系统萎缩症(MSA)、皮质基底节变性、格林-巴利(Guillain-Barre)综合征(GBS)、慢性炎性脱髓鞘性多神经病(CIDP)、癫痫症、常染色体显性遗传性夜间额叶癫痫、躁狂症、焦虑症、抑郁症、月经前烦躁不安、惊恐症、贪食症、厌食症、发作性睡病、日间过度嗜睡、双相性精神障碍、泛代性焦虑症、强迫观念与行为障碍、暴怒、对立违抗障碍、图雷特(Tourette)综合征、孤独症、药物和酒精成瘾、烟草成瘾、肥胖症、恶病质、银屑病、狼疮、急性胆管炎、口疮性口炎、溃疡、哮喘、溃疡性结肠炎、炎性肠病、局限性回肠炎、术后肠梗阻、痉挛性张力障碍、腹泻、便秘、隐窝炎、胰腺炎、病毒性肺炎、关节炎、包括类风湿性关节炎和骨关节炎、内毒素血症、脓毒病、动脉粥样硬化、特发性肺纤维化、急性疼痛、慢性疼痛、神经病变、尿失禁、糖尿病和瘤形成。认知损伤或功能障碍可能与诸如以下的精神疾病或病况有关:精神分裂症和其它精神病,包括但不限于,精神病,精神分裂症样障碍,分裂情感性障碍,妄想性障碍,短暂性精神病,共有型精神病和由一种或多种一般医学病况所引起的精神病,痴呆,以及其它认知障碍,包括但不限于轻度认知损伤,早老性痴呆,阿尔茨海默病,老年性痴呆,阿尔茨海默病型痴呆,年龄相关性记忆损伤,莱维小体痴呆,血管性痴呆,AIDS痴呆综合征,诵读困难,包括帕金森病的帕金森神经功能障碍,帕金森病的认知损伤和痴呆,多发性硬化的认知损伤,由创伤性脑损伤所引起的认知损伤,由其它的一般医学病况所引起的痴呆,焦虑症,包括但不限于,无广场恐怖症的惊恐症,伴有广场恐怖症的惊恐症,无惊恐症史的广场恐怖症,特异恐怖症,社交恐怖症,强迫观念与行为障碍,创伤后精神紧张性障碍,急性精神紧张性障碍,泛化性焦虑症和由一般医学病况所引起的泛化性焦虑症,心境障碍,包括但不限于重性抑郁障碍,心境恶劣性障碍,双相性抑郁,双相性躁狂,I型双相性精神障碍,伴随躁狂的抑郁症,抑郁或混合性发作,II型双相性精神障碍,循环性障碍和由一般医学病况所引起的心境障碍,睡眠障碍,包括但不限于睡眠障碍,原发性失眠,原发性嗜睡,发作性睡病,深眠障碍,恶梦障碍,睡眠惊恐障碍和梦游障碍,脑力发育迟缓,学习障碍,运动技能障碍,交流障碍,广泛性发育障碍,注意缺陷和分裂行为障碍,注意缺陷障碍,注意涣散多动症,婴儿、儿童或成人的进食和摄食障碍,痉挛障碍,排泄障碍,物质相关障碍,包括但不限于物质依赖,物质滥用,物质中毒,物质戒除,酒精相关障碍,安非他明或安非他明样相关障碍,咖啡因相关障碍,大麻相关障碍,可卡因相关障碍,迷幻剂相关障碍,吸入剂相关障碍,烟碱相关障碍,鸦片样物质相关障碍,苯环利定或苯环利定样相关障碍,以及镇静药、催眠要或抗焦虑药相关障碍,人格障碍,包括但不限于,强迫观念与行为人格障碍和冲动-控制障碍。精神分裂症的症状一般被分为三类:阳性,阴性和认知症状。阳性症状,有时可被称为“精神病性”症状,并包括错觉和幻觉。“阳性”是指具有明显的症状。阴性症状包括情绪平淡或缺乏表达,不能开始或完成活动,言语简短和缺少内容,以及缺乏活动的愉快感或不兴趣。“阴性”是指缺乏其它在健康受试者中存在的某些特征。认知症状属于思维过程。认知症状包括认知缺陷,诸如包括工作记忆的记忆,执行功能,注意力,警惕性,信息处理和学习,参见Sharma等人的CognitiveFunctioninSchizophrenia:Deficits,FunctionalConsequences,andFutureTreatment,Psychiatr.Clin.N.Am.26(2003)25-40,在此通过引用将其并入本说明书。精神分裂症还影响情绪。尽管许多罹患精神分裂症的受试者变得抑郁,但是一些受试者具有明显的情绪波动甚至具有双相样状态。上述的病况和障碍在例如以下文献中进一步被详细讨论:theAmericanPsychiatricAssociation:DiagnosticandStatisticalManualofMentalDisorders,第4版,TextRevision,Washington,DC,AmericanPsychiatricAssociation,2000;关于定义这些病况和障碍,通过引用将该参考文献并入本说明书。还可参见该手册关于与物质使用、滥用和依赖有关的症状和诊断特征的详细描述。优选地,进行疾病、障碍和病况的治疗或预防,而无明显的不良副作用,包括例如血压和心率的显著升高,对胃肠道的显著的不利影响,以及对骨骼肌的显著影响。本发明的化合物,当以有效量被使用时,被认为调节α7NNR的活性,而与人神经节所特有的烟碱样亚型无明显的相互作用(通过在肾上腺嗜铬组织内缺乏引发烟碱样功能的能力来证明)或与骨骼肌所特有的烟碱样亚型无明显的相互作用(通过在表达肌肉型烟碱样受体的细胞制备物中内缺乏引发烟碱样功能的能力来证明)。因此,这些化合物被认为能够治疗或预防疾病、障碍和病况,而不引发与在神经节和神经肌肉部位处的活性有关的显著的副作用。因此,化合物的给药被认为提供了治疗窗,在该治疗窗内,提供了对某些疾病、障碍和病况的治疗,并且避免了某些副作用。也就是说,有效剂量的化合物被认为足以为障碍、障碍或病况提供所需的效果,但是认为不足以(即,不在足够高的水平下)提供不希望的副作用。因此,本发明提供了本发明的化合物或其药学上可接受的盐在治疗中,诸如上述治疗中的应用。在本发明的又一个方面,提供了本发明的化合物或其药学上可接受的盐在制备用于治疗CNS障碍诸如上文所述的障碍、疾病或病况的药物中的用途。炎症已知神经系统(主要通过迷走神经)通过抑制巨噬细胞肿瘤坏死因子(TNF)的释放来调节先天免疫应答的量级。这一生理机制被称为“胆碱能抗炎途径”(例如,参见Tracey,“Theinflammatoryreflex,”Nature420:853-9(2002),通过引用将其并入本说明书)。过量的炎症因子和肿瘤坏死因子合成导致各种疾病的发病,甚至是死亡。这些疾病包括但不限于内毒素血症,类风湿性关节炎,骨关节炎,银屑病,哮喘,动脉粥样硬化,特发性肺纤维化和炎性肠病。可通过给予本说明书所述的化合物来进行治疗或预防的炎性病况包括但不限于:慢性和急性炎症,银屑病,内毒素血症,痛风,急性假性痛风,急性痛风性关节炎,关节炎,类风湿性关节炎,骨关节炎,多肌炎,皮肤肌炎,强直性脊椎炎,斯提耳(Still)病,成年发病性斯提耳病,同种异体移植排斥,慢性移植排斥,哮喘,动脉粥样硬化,单核细胞-巨噬细胞依赖性肺损伤,特发性肺纤维化,特应性皮炎,慢性阻塞性肺病,成人呼吸窘迫综合征,在镰状细胞病中的急性胸综合征,炎性肠病,局限性回肠炎,溃疡性结肠炎,急性胆管炎,口疮性口炎,隐窝炎,肾小球肾炎,狼疮肾炎,血栓形成和移植物抗宿主反应。与细菌和/或病毒感染有关的炎性应答许多的细菌和/或病毒感染(例如,脑膜炎、肝炎和肾炎)与通过毒素的形成、身体对细菌或病毒和/或毒素的自然反应所引起的副作用有关。如上所述,身体对感染的反应通常牵涉产生大量的TNF和/或其它细胞因子。这些细胞因子的过量表达可导致严重损伤,诸如脓毒性休克(当细菌是脓毒病时),内毒素性休克,尿脓毒病和中毒性休克综合征。细胞因子表达由NNR介导,并且可通过给予这些受体的激动剂或部分激动剂而受到抑制。因此,作为这些受体的激动剂或部分激动剂的本说明书所述的这些化合物可用于使得与细菌感染以及与病毒和真菌感染有关的炎性应答最小化。这些细菌感染的实例包括炭疽、肉毒中毒和脓毒病。这些化合物中的一些还具有抗微生物性质。这些化合物还可作为与目前用于处置细菌、病毒和真菌感染的治疗剂诸如抗生素、抗病毒药和抗真菌药相组合的辅助治疗剂。抗毒素也可用于结合至由感染因子所产生的毒素并使得被结合的毒素穿行通过身体而不产生炎性应答。抗毒素的实例被公开在例如Bundle等人的美国专利第6,310,043号中,通过引用将其并入本说明书。其它有效对抗细菌和其它毒素的药剂可以是有效的并且它们的治疗效果可通过与本说明书所述的化合物进行共同给药而得以互补。疼痛化合物可被给药用于治疗和/或预防疼痛,包括急性疼痛、神经学疼痛、炎性疼痛、神经性疼痛和慢性疼痛。本说明书所述的化合物的止痛活性可以在持续性炎性疼痛和神经性疼痛模型中得以证明,这些模型如美国专利申请公开文本第20010056084A1号(Allgeier等人)中所述进行,通过引用将其并入本说明书,其中在炎性疼痛的完全弗氏(Freund)佐剂大鼠模型中表现痛觉过敏和在神经性疼痛的小鼠部分坐骨神经结扎模型中表现机械痛觉过敏。止痛效果适用于治疗由各种原因或病因所导致的疼痛,特别用于治疗炎性疼痛和有关的痛觉过敏,神经性疼痛和有关的痛觉过敏,慢性疼痛(例如,严重慢性疼痛,手术后疼痛,以及与包括癌、绞痛、肾或胆绞痛、月经、偏头痛和痛风在内的各种病况有关的疼痛)。炎性疼痛可由不同的原因引起,包括关节炎和类风湿性病,腱滑膜炎和脉管炎。神经性疼痛包括三叉神经性神经痛或疱疹性神经痛,糖尿病性神经病性疼痛,灼性神经痛,下背痛,以及传入神经阻滞综合征诸如臂丛撕脱。新血管形成α7NNR与新血管形成有关。新血管形成的抑制,例如,通过给予α7NNR的拮抗剂(或在某些剂量下的部分激动剂),可以治疗或预防以不希望的新血管形成或血管发生为特征的病况。这些病况可包括以炎性血管发生和/或由缺血诱导的血管发生为特征的那些。与瘤生长有关的新血管形成也可通过给予本说明书所述的起到α7NNR的拮抗剂或部分激动剂的作用的化合物得以抑制。α7NNR特异性活性的特异性拮抗减少对炎症、缺血和瘤形成的血管发生应答。关于用于评价本说明书所述化合物的合适的动物模型系统的指导,可参见例如Heeschen,C.等人的"Anovelangiogenicpathwaymediatedbynon-neuronalnicotinicacetylcholinereceptors,"J.Clin.Invest.110(4):527-36(2002),关于血管发生的α7-特异抑制和与人类疾病有关的血管发生活性的细胞(体外)和动物模建,特别是关于Lewis肺肿瘤模型(体内,在小鼠中,特别参见第529和532-533页),通过引用将其并入本说明书。可使用本说明书所述的化合物进行治疗的代表性的肿瘤类型包括NSCLC,卵巢癌,胰腺癌,乳癌,结肠癌,直肠癌,肺癌,口咽癌,下咽癌,食管癌,胃癌,胰腺癌,肝癌,胆囊癌,胆管癌,小肠癌,尿道癌,肾癌,膀胱癌,泌尿道上皮癌,女性生殖道癌,子宫颈癌,子宫癌,卵巢癌,绒膜癌,妊娠期滋养层疾病,男性生殖道癌,前列腺癌,精囊腺癌,睾丸癌,生殖细胞瘤,内分泌腺癌,甲状腺癌,肾上腺癌,脑下垂体癌,皮肤癌,血管瘤,黑素瘤,肉瘤,骨和软组织肉瘤,卡波西肉瘤,脑瘤,神经瘤,眼瘤,脑膜瘤,星形细胞瘤,神经胶质瘤,成胶质细胞瘤,视网膜母细胞瘤,神经瘤,神经母细胞瘤,神经鞘瘤,脑膜瘤,由造血恶性病引起的实体瘤(诸如白血病,绿色瘤,浆细胞瘤,以及蕈样真菌病的斑块和肿瘤,以及皮肤T细胞淋巴瘤/白血病),以及得自淋巴瘤的实体瘤。所述化合物还可与其它形式的抗癌疗法组合使用,包括与抗肿瘤药诸如顺铂、阿霉素、柔红霉素等共同给药,以及/或与抗-VEGF(血管内皮细胞生长因子)药剂共同给药,这些是本领域已知的。化合物可以使它们靶向于肿瘤部位的方式被给药。例如,化合物可以在与各种抗体缀合的微球、微粒或脂质体内被给药,所述抗体引导所述的微粒子到达肿瘤。另外,化合物可以存在于这样的微球、微粒或脂质体内,这些微球、微粒或脂质体经过适当地筛分以穿行通过动脉和静脉、但进入并固定在肿瘤周围的毛细管床内,并将所述化合物局部给予到肿瘤。这些药物递送装置是本领域已知的。其它障碍除了治疗CNS障碍、炎症、新血管形成和疼痛之外,本发明的化合物还可用于预防或治疗某些其它的其中NNR起作用的病况、疾病和障碍。实例包括自身免疫障碍诸如狼疮,与细胞因子释放有关的障碍,继发于感染的恶病质(例如,在AIDS、AIDS相关综合征和瘤形成中存在的),代谢障碍,包括I型糖尿病,II型糖尿病,代谢综合征,肥胖症或高血糖症,pemphitis,尿失禁,视网膜疾病,感染性疾病,肌无力,类重症肌无力(Eaton-Lambert)综合征,高血压,骨质疏松症,血管收缩,血管舒张,心律不齐,贪食症,厌食症以及在公开的PCT申请WO98/25619中所述的那些适应症,关于这些障碍,将该文献并入本说明书。本发明的化合物还可被给予以治疗惊厥,诸如作为癫痫症状的惊厥,并用于治疗诸如梅毒和克-雅病等病况。诊断应用所述化合物可用于诊断用组合物,诸如探针,特别是当它们经过修饰以包括合适的标记物时。探针可用于例如确定特异性受体,特别是α7受体亚型的相对数目和/或功能。为此目的,本发明的化合物最优选用放射性同位素部分诸如11C、18F、76Br、123I或125I进行标记。被给予的化合物可采用适于所用标记的已知的检测方法进行检测。检测方法的实例包括正电子发射断层摄影术(PET)和单光子发射计算机断层摄影术(SPECT)。如上所述的放射性标记物可用于PET(例如,11C,18F或76Br)和SPECT(例如,123I)成像中,11C的半衰期为约20.4分钟,18F的半衰期为约109分钟,123I的半衰期为约13小时,以及76Br的半衰期为约16小时。希望高的比放射性以使得在非饱和浓度下的被选择的受体亚型显像。被给予的剂量典型地低于毒性剂量并提供强对比图象。化合物预计能够在无毒水平下被给予。以放射性标记成像领域技术人员已知的方式来确定剂量。例如,参见London等人的美国专利第5,969,144号,关于给予这种化合物,通过引用将其并入本说明书。所述化合物可以使用已知的技术被给予。例如,参见London等人的美国专利第5,969,144号,关于这种给药,通过引用将其并入本说明书。化合物可在并入其它成分(诸如可用于配制诊断组合物的那些类型的成分)的制剂组合物中被给予。根据本发明可使用的化合物最优选以高纯度的形式被使用。参见Elmalch等人的美国专利第5,853,696号,关于这种分析,通过引用将其并入本说明书。在化合物被给予到受试者(例如人受试者)后,在受试者内存在的化合物可以通过合适的技术进行成像和定量化,以便指示被选择的NNR亚型的存在、量和功能。除了人之外,化合物也可被给予到动物,诸如小鼠、大鼠、狗和猴。SPECT和PET成像可使用任何合适的技术和装置来进行。参见Villemagne等人,Arneric等人(编)的NeuronalNicotinicReceptors:PharmacologyandTherapeuticOpportunities,235-250(1998)和Elmalch等人的美国专利第5,853,696号中,关于代表性成像技术,通过引用将它们各自并入本说明书。放射性标记的化合物以高亲和性结合于选择性NNR亚型(例如α7),并优选表现出很小的与其它烟碱性胆碱能受体亚型(例如,与肌肉和神经节有关的那些受体亚型)的非特异性结合。就这点而论,所述化合物可用作用于受试者体内的烟碱性胆碱能受体亚型的非侵害性成像的试剂,特别地用在脑内用于与各种CNS疾病和障碍有关的诊断。在一个方面,诊断组合物可用于诊断受试者诸如人患者的疾病的方法中。所述方法涉及对患者给予经过可检测标记的本说明书所述的化合物,并检测该化合物与被选择的NNR亚型(例如,α7受体亚型)的结合。使用诊断工具诸如PET和SPECT的本领域技术人员可以使用本说明书所述的放射性标记化合物来诊断各种病况和障碍,包括与中枢神经系统和自主神经系统的功能障碍有关的病况和障碍。这些障碍包括多种CNS疾病和障碍,包括阿尔茨海默病,帕金森病和精神分裂症。可进行评价的这些和其它的代表性疾病和障碍包括在Bencherif等人的美国专利5,952,339中所述的那些,该文献在此通过引用将其并入本说明书。在另一个方面,诊断组合物可用于监控受试者诸如人患者的选择性烟碱样受体亚型的方法中。所述方法涉及对患者给予经过可检测标记的本说明书所述的化合物,并检测该化合物与被选择的烟碱样受体亚型即α7受体亚型的结合。受体结合本发明的化合物在与NNR亚型、特别是α7受体亚型结合的化合物的结合试验中用作参比配体。为此目的,本发明的化合物优选用放射性同位素部分诸如3H或14C进行标记。V.合成实施例提供了以下的合成实施例来说明本发明而不认为该实施例对本发明的范围构成限制。在这些实施例中,所有的份数和百分数都以重量计,除非另作说明。所有的溶液是含水的,除非另作说明。实施例1:小规模合成(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(化合物A)及其对映体(2R,3S)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺2-((3-吡啶基)亚甲基)-1-氮杂双环[2.2.2]辛烷-3-酮将氢氧化钾(56g,0.54摩尔)溶解在甲醇(420mL)中。加入3-奎宁环酮盐酸盐(75g,0.49摩尔)并将混合物在周围环境温度搅拌30分钟。加入3-吡啶甲醛(58g,0.54摩尔)并将混合物在周围环境温度搅拌16小时。反应混合物在此期间变成黄色,有固体粘结在烧瓶壁上。从壁上刮下固体并使其破碎。在快速搅拌下,加入水(390mL)。当固体溶解时,将混合物在4℃冷却过夜。通过过滤收集晶体,用水洗涤,并空气干燥,得到80g的黄色固体。通过将滤液浓缩到其原来体积的约10%并在4℃冷却过夜得到第二批(8g)产物。两批具有足够的用于进一步转化的纯度(88g,82%收率)。2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮将2-((3-吡啶基)亚甲基)-1-氮杂双环[2.2.2]辛烷-3-酮(20g,93mmol)悬浮在甲醇(200mL)中并用46mL的6M盐酸处理。加入10%的炭载钯(1.6g)并将混合物在25psi氢气下振摇16小时。将混合物过滤通过硅藻土,并通过旋转蒸发除去滤液中的溶剂。得到粗制2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮盐酸盐,为白色胶状物(20g),将其随后用2M氢氧化钠(50mL)和氯仿(50mL)处理并搅拌1小时。分离氯仿层,将水相用2M氢氧化钠(~5mL,足够使pH升高到10)和饱和的氯化钠水溶液(25mL)处理。将该含水混合物用氯仿(3×10mL)提取,将合并的氯仿提取物干燥(无水硫酸镁)并通过旋转蒸发进行浓缩。将残余物(18g)溶解在温热的醚(320mL)中并冷却到4℃。滤出该白色固体,用少量冷乙醚洗涤并空气干燥。将滤液浓缩到其原来体积的约10%并在4℃冷却,得到第二批。合并收率为16g(79%)。3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷在氮气下,向搅拌的2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮(3.00g,13.9mmol)在无水甲醇(20mL)中的溶液中加入1M的氯化锌在醚中的溶液(2.78mL,2.78mmol)。在周围环境温度搅拌30分钟后,将该混合物用固体甲酸铵(10.4g,167mmol)处理。在周围环境温度搅拌另外1小时后,分份加入固体氰基硼氢化钠(1.75g,27.8mmol)。然后将反应在周围环境温度搅拌过夜并通过加入水(~5mL)使反应终止。将经淬灭的反应在5M氢氧化钠(10mL)和氯仿(20mL)之间分配。水层用氯仿(20mL)提取,并将合并的有机层干燥(硫酸钠),过滤并浓缩。得到2.97g的黄色胶状物。GCMS分析显示该产物是顺式胺和反式胺的1:9的混合物,并一起存在痕量的相应的醇(98%的总质量回收率)。(2R,3S)和(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷将二-对甲苯酰基-D-酒石酸(5.33g,13.8mmol)加入到粗制3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷(6.00g,27.6mmol的1:9的顺式/反式混合物)在甲醇(20mL)中的溶液中。完全溶解后,通过旋转蒸发将澄清溶液浓缩形成固体物质。将固体溶解在最小量的煮沸甲醇(~5mL)中。将溶液缓慢地冷却,首先冷却到周围环境温度(1小时),然后在5℃历时约4小时,并最后在-5℃过夜。通过抽滤收集沉淀的盐,并从5mL的甲醇重结晶。空气干燥得到1.4g的白色固体,将其在氯仿(5mL)和2M氢氧化钠(5mL)之间分配。氯仿层和水层的5mL氯仿提取物被合并,干燥(无水硫酸钠)并浓缩,得到无色的油状物(0.434g)。该游离碱的对映体纯度通过将一部分转化成其N-(叔丁氧羰基)-L-脯氨酰胺、然后使用LCMS分析非对映纯度(98%)来测定。用2M氢氧化钠使从最初结晶得到的母液呈碱性(~pH11)并用氯仿(10mL)提取两次。将氯仿提取物干燥(无水硫酸钠)并浓缩,得到油状物。将该胺(3.00g,13.8mmol)溶解在甲醇(10mL)中并用二-对甲苯酰基-L-酒石酸(2.76g,6.90mmol)处理。将混合物温热以帮助溶解,然后缓慢地冷却到-5℃,在-5℃将其保持过夜。通过抽滤收集沉淀物,从甲醇重结晶并干燥。得到1.05g的白色固体。将该盐转化成游离碱(收率=0.364g),并如上文关于另一个对映体所述,使用脯氨酰胺方法测定对映体纯度(97%)。N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺的反式对映体A将氯磷酸二苯酯(0.35mL,0.46g,1.7mmol)滴加到苯并呋喃-2-甲酸(0.28g,1.7mmol)和三乙胺(0.24mL,0.17g,1.7mmol)在无水二氯甲烷(5mL)中的溶液中。在周围环境温度搅拌30分钟后,加入(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷(0.337g,1.55mmol)(其衍生自二-对甲苯酰基-D-酒石酸盐)和三乙胺(0.24mL,0.17g,1.7mmol)在无水二氯甲烷(5mL)中的溶液。将反应混合物在周围环境温度搅拌过夜,然后用10%氢氧化钠(1mL)处理。分离双相混合物,在Genevac离心蒸发器上浓缩有机层。将残余物溶解在甲醇(6mL)中并在C18硅胶柱上,使用含0.05%三氟乙酸的乙腈/水梯度作为洗脱液进行HPLC纯化。浓缩所选择的级分,将得到的残余物在氯仿和饱和碳酸氢钠水溶液之间分配,并蒸发氯仿,得到0.310g(42%收率)的白色粉末(GCMS纯度为95%)。1HNMR(300MHz,CDCl3)δ8.51(d,1H),8.34(dd,1H),7.66(d,1H),7.58(dt,1H),7.49(d,1H),7.44(s,1H),7.40(dd,1H),7.29(t,1H),7.13(dd,1H),6.63(d,1H),3.95(t,1H),3.08(m,1H),2.95(m,4H),2.78(m,2H),2.03(m,1H),1.72(m,3H),1.52(m,1H)。随后通过手性色谱分析,确定该物质(反式对映体A)与绝对构型是2S,3R(通过X射线结晶学分析确定)的物质相同。N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺的反式对映体B将氯磷酸二苯酯(96μL,124mg,0.46mmol)滴加到苯并呋喃-2-甲酸(75mg,0.46mmol)和三乙胺(64μL,46mg,0.46mmol)在无水二氯甲烷(1mL)中的溶液中。在周围环境温度搅拌45分钟后,加入(2R,3S)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷(0.10g,0.46mmol)(其衍生自二-对甲苯酰基-L-酒石酸盐)和三乙胺(64μL,46mg,0.46mmol)在无水二氯甲烷(1mL)中的溶液中。将反应混合物在周围环境温度搅拌过夜,然后用10%氢氧化钠(1mL)处理。分离双相混合物,有机层和水层的氯仿提取物(2mL)通过旋转蒸发进行浓缩。将残余物溶解在甲醇中并在C18硅胶柱上,使用含有0.05%三氟乙酸的乙腈/水梯度作为洗脱液进行HPLC纯化。浓缩所选择的级分,将得到的残余物在氯仿和饱和碳酸氢钠水溶液之间分配,并蒸发氯仿,得到82.5mg(50%收率)的白色粉末。NMR谱与2S,3R异构体的NMR谱相同。因为该物质(反式对映体B)的直接前体与2S,3R化合物(反式对映体A)的直接前体是对映体关系,因此反式对映体B的绝对构型被假定为是2R,3S。实施例2:大规模合成(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺和(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)-1-苯并呋喃-2-甲酰胺对甲苯磺酸盐2-((3-吡啶基)亚甲基)-1-氮杂双环[2.2.2]辛烷-3-酮在氮气气氛下,将3-奎宁环酮盐酸盐(8.25kg,51.0mol)和甲醇(49.5L)加入到100L的玻璃反应烧瓶中,所述反应烧瓶装备有机械搅拌器、测温探头和冷凝器。通过添粉漏斗将氢氧化钾(5.55kg,99.0mol)在约30分钟内加入,导致反应温度从50℃升高到56℃。在约2小时内,将3-吡啶甲醛(4.80kg,44.9mol)加入到反应混合物中。将得到的混合物在20℃±5℃搅拌至少12小时,并通过薄层色谱法(TLC)监控反应。反应完成后,将反应混合物过滤通过烧结玻璃漏斗并将滤饼用甲醇(74.2L)洗涤。将滤液浓缩,转移到反应烧瓶中,并加入水(66.0L)。将悬浮液搅拌至少30分钟,过滤,并将滤饼用水(90.0L)洗涤直到洗液的pH为7-9。将固体在50℃±5℃下真空干燥至少12小时,得到8.58kg(89.3%)的2-((3-吡啶基)亚甲基)-1-氮杂双环[2.2.2]辛烷-3-酮。(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮二-对甲苯酰基-D-酒石酸盐在惰性气氛下,将2-((3-吡啶基)亚甲基)-1-氮杂双环[2.2.2]辛烷-3-酮(5.40kg,25.2mol)和甲醇(40.5L)加入到72L反应容器中,该反应容器装备有机械搅拌器、测温探头、低压气体调节系统和压力计。将顶部空间充满氮气,并将混合物搅拌以获得澄清的黄色溶液。向该烧瓶中加入10%的炭载钯(50%湿度)(270g)。使用真空泵将反应器的气氛排空,并用氢气置换顶部空间达到10-20英寸的水压力。排气和用氢气加压再重复2次,使得在第三次加压后反应器处于20英寸的水压力下的氢气中,将反应混合物在20℃±5℃搅拌至少12小时,并通过TLC监控反应。反应完成后,将悬浮液过滤通过在烧结玻璃漏斗上的545垫(1.9kg),并将滤饼用甲醇(10.1L)洗涤。将滤液浓缩得到半固体,在氮气气氛下将该半固体转移到200L反应烧瓶中,该反应烧瓶装备有机械搅拌器、冷凝器和测温探头。将该半固体溶解在乙醇(57.2L)中,并加入二-对甲苯酰基-D-酒石酸(DTTA)(9.74kg,25.2mol)。将搅拌的反应混合物在回流下加热至少1小时,将反应保持另外的至少12小时并同时将反应冷却到15℃到30℃。使用桌面过滤器将悬浮液过滤,将滤饼用乙醇(11.4L)洗涤。将产品在周围环境温度下真空干燥,得到11.6kg(76.2%收率,59.5%的纯度因数)的(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮二-对甲苯酰基-D-酒石酸盐。(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷二-对甲苯酰基-D-酒石酸盐将水(46.25L)和碳酸氢钠(4.35kg,51.8mol)加入到200L烧瓶中。完全溶解后,加入二氯甲烷(69.4L)。加入(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮二-对甲苯酰基-D-酒石酸盐(11.56kg,19.19mol),并将反应混合物搅拌2-10分钟。使层分离历时至少2分钟(当必要时加入另外的水(20L)以分层)。除去有机相并用无水硫酸钠干燥。向剩余的水相中加入二氯甲烷(34.7L),并将悬浮液搅拌2-10分钟。使层分离,历时2-10分钟。再次,除去有机相并用无水硫酸钠干燥。再次如上所述重复用二氯甲烷(34.7L)提取水相。每次提取的样品用于手性HPLC分析。通过过滤除去硫酸钠,并将滤液浓缩,得到(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮(4.0kg),为固体。在氮气气氛下,将(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮(3.8kg)转移到洁净的100L玻璃反应烧瓶中,该反应烧瓶装备有机械搅拌器和测温探头。加入无水四氢呋喃(7.24L)和(+)-(R)-α-甲基苄基胺(2.55L,20.1mol)。将异丙醇钛(IV)(6.47L,21.8mol)在1小时内加入到搅拌的反应混合物中。将反应在氮气气氛下搅拌至少12小时。将乙醇(36.17L)加入到反应混合物中。将反应混合物冷却到低于-5℃,并分份加入硼氢化钠(1.53kg,40.5mol),保持反应温度低于15℃(这一加料需要几个小时)。然后将反应混合物在15℃±10℃搅拌至少1小时。通过HPLC监控反应,反应完成(由剩余低于0.5%的(2S)-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-酮所显示)后,加入2M氢氧化钠(15.99L)并将混合物搅拌至少10分钟。将反应混合物过滤通过桌面漏斗内的545垫。将滤饼用乙醇(15.23L)洗涤,并将滤液浓缩,得到油状物。在惰性气氛下将浓缩物转移到洁净的100L玻璃反应烧瓶中,该反应烧瓶装备有机械搅拌器和测温探头。加入水(1L),并将混合物冷却到0℃±5℃。将2M盐酸(24L)加入到混合物中以调节混合物的pH到pH1。然后将混合物搅拌至少10分钟,并缓慢地加入2M氢氧化钠(24L)以调节混合物的pH到pH14。将混合物搅拌至少10分钟,将水相用二氯甲烷(3×15.23L)提取。将有机相用无水硫酸钠(2.0kg)干燥,过滤并浓缩,得到(2S,3R)-N-((1R)-苯基乙基)-3-氨基-2-((3-吡啶基)甲基))-1-氮杂双环[2.2.2]辛烷(4.80kg,84.7%收率)。在惰性气氛下,将(2S,3R)-N-((1R)-苯基乙基)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷转移到22L的玻璃烧瓶中,该烧瓶装备有机械搅拌器和测温探头。加入水(4.8L),并将搅拌的混合物冷却到5℃±5℃。将浓盐酸(2.97L)缓慢地加入到反应烧瓶中,保持混合物的温度低于25℃。在惰性气氛下,将得到的溶液转移到含有乙醇(18L)并装备有机械搅拌器、测温探头和冷凝器的72L反应烧瓶中。向该烧瓶中加入10%的炭载钯(50%湿度)(311.1g)和环己烯(14.36L)。将反应混合物在接近回流的温度加热至少12小时,并通过TLC监控反应。反应完成后,将反应混合物冷却到低于45℃,将其过滤通过在烧结玻璃漏斗上的545垫(1.2kg)。将滤饼用乙醇(3L)漂洗并将滤液浓缩,得到水相。将水(500mL)加入到经过浓缩的滤液中,并将该合并的水层用甲基叔丁基醚(MTBE)(2x4.79L)洗涤。将2M氢氧化钠(19.5L)加入到水相中以调节混合物的pH到pH14。然后将混合物搅拌至少10分钟。将水相用氯仿(4×11.96L)提取,将合并的有机相用无水硫酸钠(2.34kg)干燥。将滤液过滤并浓缩,得到(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷(3.49kg,大于定量收率),为油状物。在惰性气氛下,将(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷转移到洁净的100L反应烧瓶中,该反应烧瓶装备有机械搅拌器、冷凝器和测温探头。加入乙醇(38.4L)和二-对甲苯酰基-D-酒石酸(3.58kg,9.27mol)。将反应混合物在温和回流下加热至少1小时。然后将反应混合物搅拌至少12小时同时使其冷却到15℃到30℃。将得到的悬浮液过滤,将滤饼用乙醇(5.76L)洗涤。在惰性气氛下,将滤饼转移到洁净的100L玻璃反应烧瓶中,该反应烧瓶装备有机械搅拌器、测温探头和冷凝器。加入9:1的乙醇/水溶液(30.7L),将得到的浆料在温和回流下加热至少1小时。然后将反应混合物搅拌至少12小时同时使其冷却到15℃到30℃。将混合物过滤器并将滤饼用乙醇(5.76L)洗涤。收集产品并在50℃±5℃真空干燥至少12小时,得到5.63kg(58.1%收率)的(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷二-对甲苯酰基-D-酒石酸盐。(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺在惰性气氛下,将(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷二-对甲苯酰基-D-酒石酸盐(3.64kg,5.96mol)和10%的氯化钠水溶液(14.4L,46.4mol)加入到装备有机械搅拌器的72L的玻璃反应烧瓶中。将5M氢氧化钠(5.09L)加入到搅拌的混合物中以调节混合物的pH到pH14。然后将混合物搅拌至少10分钟。将含水溶液用氯仿(4x12.0L)提取,将合并的有机层用无水硫酸钠(1.72kg)干燥。将合并的有机层过滤,将滤液浓缩,得到(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷(1.27kg),为油状物。在惰性气氛下,将(2S,3R)-3-氨基-2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷转移到装备有机械搅拌器的50L玻璃反应烧瓶中。将二氯甲烷(16.5L)、三乙胺(847mL,6.08mol)、苯并呋喃-2-甲酸(948g,5.85mol)和O-(苯并三唑-1-基)-N,N,N,1-四甲基铵六氟磷酸盐(HBTU)(2.17kg,5.85mol)加入到反应混合物中。将混合物在周围环境温度搅拌至少4小时,并通过HPLC监控反应。反应完成后,将10%的碳酸钾水溶液(12.7L,17.1mol)加入到反应混合物中并将混合物搅拌至少5分钟。分层并将有机相用10%的盐水(12.7L)洗涤,分层并将有机相冷却到15℃±10℃。将3M盐酸(8.0L)缓慢地加入到反应混合物中以调节混合物的pH到pH1。然后将混合物搅拌至少5分钟。使层分离历时至少5分钟。使用桌面过滤器对固体过滤。将滤液分层,并将水相和得自漏斗的固体转移到反应烧瓶中。将3M氢氧化钠(9.0L)分份缓慢地加入到烧瓶中以调节混合物的pH到pH14。将水相用二氯甲烷(2×16.5L)提取。将合并的有机相用无水硫酸钠(1.71kg)干燥。将混合物过滤,将滤液浓缩,得到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(1.63kg,77.0%收率),为黄色固体。(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺对甲苯磺酸盐将(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛烷-3-基)苯并呋喃-2-甲酰胺(1.62kg,4.48mol)和二氯甲烷(8.60kg)加入到小口大玻璃瓶中。通过HPLC分析测定溶液中物质的重量/重量%。将溶液浓缩形成油状物,加入丙酮(4L),将混合物浓缩形成含油固体。加入另外的丙酮(12L)到位于旋转蒸发器蒸发球中的含油固体中,在惰性气氛下将得到的浆料转移到装备有机械搅拌器、冷凝器、测温探头和冷凝器的50L玻璃反应烧瓶中。将反应混合物加热到50℃±5℃。将水(80.7g)加入溶液,并将其搅拌至少10分钟。将对甲苯磺酸(853g,4.44mol)在约15分钟内分份加入到反应混合物中。将反应混合物加热到回流并在该温度保持至少30分钟以获得溶液。将反应在约2小时内冷却到40℃±5℃。在约1.5小时内加入乙酸异丙酯(14.1L)。在至少10小时内将反应混合物缓慢地冷却到周围环境温度。将混合物过滤并将滤饼用乙酸异丙酯(3.5L)洗涤。将分离的产品在105℃±5℃真空干燥2-9小时,得到2.19kg(88.5%收率)的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺对甲苯磺酸盐,mp226-228℃。1HNMR(500MHz,D2O)δ8.29(s,1H),7.78(m,J=5.1,1H),7.63(d,J=7.9,1H),7.54(d,J=7.8,1H),7.49(d,J=8.1,2H),7.37(m,J=8.3,1H),7.33(m,J=8.3,6.9,1.0,1H),7.18(m,J=7.8,6.9,1.0,1H),7.14(d,J=8.1,2H),7.09(s,1H),6.99(dd,J=7.9,5.1,1H),4.05(m,J=7.7,1H),3.74(m,1H),3.47(m,2H),3.28(m,1H),3.22(m,1H),3.15(dd,J=13.2,4.7,1H),3.02(dd,J=13.2,11.5,1H),2.19(s,3H),2.02(m,2H),1.93(m,2H),1.79(m,1H).13CNMR(126MHz,D2O)δ157.2,154.1,150.1,148.2,146.4,145.2,138.0,137.0,130.9,128.2(2),126.9,126.8,125.5(2),123.7,123.3,122.7,111.7,100.7,61.3,50.2,48.0,40.9,33.1,26.9,21.5,20.8,17.0。通过用氢氧化钠水溶液处理并用氯仿提取,将该物质的样品转化为化合物A游离碱(用于盐选择研究)。充分蒸发氯仿产生灰白色粉末,mp167-170℃,该粉末具有以下的谱图特征:正离子电喷射MS[M+H]+离子m/z=362。1HNMR(500MHz,DMSO-d6)δ8.53(d,J=7.6Hz,1H),8.43(d,J=1.7Hz,1H),8.28(dd,J=1.6,4.7Hz,1H),7.77(d,J=7.7Hz,1H),7.66(d,J=8.5Hz,1H),7.63(dt,J=1.7,7.7Hz,1H),7.52(s,1H),7.46(m,J=8.5,7.5Hz,1H),7.33(m,J=7.7,7.5Hz,1H),7.21(dd,J=4.7,7.7Hz,1H),3.71(m,J=7.6Hz,1H),3.11(m,1H),3.02(m,1H),2.80(m,2H),2.69(m,2H),2.55(m,1H),1.80(m,1H),1.77(m,1H),1.62(m,1H),1.56(m,1H),1.26(m,1H).13CNMR(126MHz,DMSO-d6)δ158.1,154.1,150.1,149.1,146.8,136.4,135.4,127.1,126.7,123.6,122.9,122.6,111.8,109.3,61.9,53.4,49.9,40.3,35.0,28.1,26.1,19.6。化合物A的一盐酸盐(参见实施例5)用于X射线结晶学分析。得到的晶体结构(分别如图10A和10B所示)确认了化合物A的2S,3R绝对构型。实施例3:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺磷酸盐的合成向圆底烧瓶中加入(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(8.18g,22.6mmol)和2-丙醇(180mL)。将混合物在65-70℃搅拌和加热,直到所有固体溶解。将溶液在65-70℃剧烈搅拌,通过移液管滴加磷酸(1.65mL,24.3mmol)。立刻形成白色的粒状固体。将混合物在65-70℃搅拌30分钟,冷却到周围环境温度(23℃)并搅拌另外24小时。通过吸滤收集白色固体,将滤饼用2-丙醇(20mL)洗涤并将固体空气干燥至少1小时。将固体在真空箱中在75℃进一步干燥过夜(16小时),得到10.7g的产物(大于定量收率),熔点265-273℃(分解),在约180℃观察到结晶度改变。1H-NMR(DMSO-d6)显示存在2-丙醇(强溶剂合物),其可解释大于定量收率的原因。手性LC分析得出97.1%的纯度(270nm)。实施例4:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺马来酸盐的合成将马来酸(0.067g,0.630mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(0.203g,0.560mmol)在乙酸异丙酯(2mL)中的热浆料中,有细的白色固体沉淀析出,同时还有胶质残余物。加入另外的乙酸异丙酯(3mL)和马来酸(0.006g),将混合物加热至回流,在回流下加入异丙醇(5mL)。将得到的白色固体混合物冷却到周围环境温度,过滤,将固体用乙酸异丙酯(2mL)洗涤。将产物在60℃真空干燥18小时,得到0.228g的灰白色的片状固体(84.7%收率),mp180-182℃。1HNMR(DMSO-d6)显示是单盐化学计量。C22H23N3O2C4H4O4的计算值:C,65.40;H,5.70;N,8.80;实测值:C,65.35,65.29;H,5.86,5.68;N,8.69,8.78。实施例5:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺盐酸盐的合成一盐酸盐:通过将浓盐酸(1.93mL,12M,23.2mmol)滴加到8.5mL的冷THF中来制备盐酸/THF溶液。将该溶液温热到周围环境温度。向圆底烧瓶中加入(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(8.49g,23.5mmol)和丙酮(85mL)。将混合物在45-50℃搅拌和加热,直到实现完全溶解。在5分钟内滴加上面制备的盐酸/THF溶液,使用另外的THF(1.5mL)用于转移。在加入酸性溶液期间开始形成粒状的白色固体。将混合物冷却到周围环境温度,并搅拌过夜(16小时)。通过抽滤收集固体,将滤饼用丙酮(10mL)洗涤,将固体在抽吸下空气干燥30分钟。将固体在真空箱中在75℃另外干燥2小时,得到8.79g的细的白色晶体(94%收率),mp255-262℃。手性LC分析得到98.8%的纯度(270nm)。1H-NMR(DMSO-d6)显示无残留溶剂并证实单化学计量。1HNMR(300MHz,DMSO-d6)δ10.7(宽的s,1H-季铵),8.80(宽的s,1H-酰胺H),8.54(s,1H),8.23(d,1H),7.78(d,1H),7.74(d,1H),7.60(d,1H),7.47(m,2H),7.33(m,1H),7.19(m,1H),4.19(m,1H),4.08(m,1H),3.05-3.55(m,6H),2.00-2.10(m,3H),1.90(m,1H),1.70(m,1H)。该盐的X射线结晶学分析确认了立体化学归属和化学计量(参见图10A和10B)。二盐酸盐:将氯化氢气体缓慢地鼓泡通过(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(1.9g,5.3mmol)在无水乙醚(25mL)中的冰冷却的溶液中。首先在氮气气流中,然后使用高真空(在高真空管中的氢氧化钠洗涤器)除去挥发物。将残余物与小体积的无水乙醚(被废弃)研磨,并将剩余的固体高真空干燥。得到2.17g(94%收率)灰白色粉末,mp210-212℃(吸湿的)。手性LC分析得出93.7%的纯度(270nm)。正离子电喷射MS[M+H]+离子m/z=362。1HNMR(300MHz,CD3OD)δ9.15(s,1H),8.84(d,1H),8.63(d,1H),7.97(t,1H),7.75(d,1H),7.61(d,1H),7.52(m,2H),7.35(t,1H),4.50(m,1H),4.32(m,1H),3.40-3.85(m,6H),1.95-2.40(m,5H)。实施例6:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺半半乳糖二酸盐的合成将半乳糖二酸(粘酸)(36.3mg,0.173mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液中。当加入水(8滴)时将混合物回流;然后使热混合物过滤通过棉花塞,随后将其用乙醇(1mL)漂洗。冷却不能得到沉淀物。通过旋转蒸发除去挥发物,将残余物(白色泡沫)与异丙醇(被废弃)研磨,将剩余的固体溶解在回流的丙酮/水(4mL,7:1)中。缓慢冷却到5℃,产生白色固体,将其滤出,用异丙醇(3×1mL)洗涤,并高真空干燥。这样得到118mg(73%收率)的细白色片状物,mp134-139℃。1HNMR(300MHz,D2O)δ8.29(s,1H),7.78(d,1H),7.62(d,1H),7.54(d,1H),7.35(m,2H),7.18(t,1H),7.10(s,1H),6.98(m,1H),4.08(s,1H,半乳糖二酸),3.98(d,1H),3.77(s,1H,半乳糖二酸),3.66(m,1H),3.35(m,1H),2.95-3.30(m,4H),1.65-2.05(m,5H)。实施例7:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺D-酒石酸盐的合成将酒石酸(25.6mg,0.173mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液中。将得到的溶液缓慢地冷却到周围环境温度。无固体沉淀析出,因此将溶液浓缩,得到白色泡沫。在异丙醇中结晶的尝试以失败告终。将该泡沫溶解在甲醇中并加入另外一半当量的酒石酸(25.6mg,0.173mmol)。将混合物浓缩,得到白色泡沫,其不从甲醇和异丙醇的混合物中结晶。然后将经过浓缩的物质(固体和胶状液体的混合物)在乙酸乙酯(1mL)中成浆料,得到白色固体。其通过过滤进行分离(用乙酸乙酯洗涤)并在真空箱中干燥(18小时,在40℃),得到141mg(79.7%收率)的单化学计量的盐(NMR),mp136-140℃。手性LC分析得到98.1%的纯度(270nm)。1HNMR(300MHz,D2O)δ8.50(s,1H),8.01(d,1H),7.86(d,1H),7.75(d,1H),7.56(m,2H),7.38(t,1H),7.32(s,1H),7.21(t,1H),4.34(s,2H,酒石酸),4.26(d,1H),3.95(m,1H),3.64(m,2H),3.15-3.55(m,4H),1.90-2.30(m,5H)。实施例8:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺甲磺酸盐的合成将甲磺酸(33.2mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液中。冷却不能产生沉淀物。将混合物回流,并使热混合物过滤通过棉花塞,随后将其用甲醇(1mL)漂洗。通过旋转蒸发除去挥发物,并将残余物(浅黄色泡沫)溶解在热异丙醇(1mL)中。再次,冷却不能产生沉淀物。蒸发异丙醇,并将残余物在丙酮(1mL)中成浆料。过滤并在真空箱中干燥(18小时,在50℃),得到146mg(92.5%收率)的浅米色固体,mp240-243℃。1HNMR(300MHz,D2O)δ8.32(s,1H),7.82(d,1H),7.66(d,1H),7.57(d,1H),7.38(m,2H),7.20(m,1H),7.12(s,1H),7.01(m,1H),4.09(d,1H),3.75(m,1H),3.47(m,2H),3.00-3.40(m,4H),2.67(s,3H,甲磺酸),1.75-2.15(m,5H)。实施例9:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺D-扁桃酸盐的合成将D-扁桃酸(52.6mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液中。用乙酸乙酯(4ml)稀释并冷却不能产生沉淀物。通过旋转蒸发除去挥发物,并将残余物(白色泡沫)溶解在热异丙醇(0.5mL)中。冷却到5℃,产生白色结晶,通过抽滤收集该结晶。真空箱干燥(18小时,在45℃),得到111mg(62.4%收率)的浅米色固体,mp188.5-193℃。1HNMR(300MHz,D2O)δ8.33(s,1H),7.83(s,1H),7.67(d,1H),7.60(d,1H),7.27(m,8H,包含扁桃酸),7.12(s,1H),7.01(m,1H),4.85(s,1H,扁桃酸),4.10(d,1H),3.75(m,1H),3.48(m,2H),3.00-3.40(m,4H),1.75-2.15(m,5H)。实施例10:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺R-樟脑磺酸盐的合成将R-10-樟脑磺酸(80.3mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液中。冷却不能沉积任何沉淀物。通过旋转蒸发除去挥发物,并将残余物(白色泡沫)溶解在热异丙醇(0.5mL)中。冷却到5℃产生很少的白色结晶和乳状悬浮液。用刮铲刮擦烧瓶侧壁使混合物最终转化成细白色结晶的厚块。加入另外0.5mL的异丙醇,通过抽滤收集结晶。真空箱干燥(在70℃历时5小时,然后在110℃历时2小时),得到193mg(93.8%收率)的白色固体。mp149.5-156℃。1HNMR(300MHz,D2O)δ8.30(s,1H),7.79(d,1H),7.64(d,1H),7.55(d,1H),7.36(m,2H),7.18(m,1H),7.11(s,1H),6.99(m,1H),4.07(d,1H),3.73(m,1H),3.45(m,2H),3.95-3.35(m,5H,包含樟脑磺酸),2.64(d,1H,樟脑磺酸),2.22(m,2H),1.70-2.10(m,8H,包含樟脑磺酸),1.45(m,1H,樟脑磺酸),1.25(m,1H,樟脑磺酸),0.85(s,3H,樟脑磺酸),0.68(s,3H,樟脑磺酸)。实施例11:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺S-樟脑磺酸盐的合成将S-10-樟脑磺酸(80.3mg,0.346mmol)加入到(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺(125mg,0.346mmol)在热乙醇(1mL)中的溶液。用乙酸乙酯(4mL)稀释,冷却不能沉积任何沉淀物。通过旋转蒸发除去挥发物,并将残余物(白色泡沫)溶解在热异丙醇(1.5mL)中。冷却到5℃,产生白色结晶。将混合物浓缩到约0.5mL并再次冷却到5℃。然后通过抽滤收集固体并最初在45℃真空干燥18小时,然后在持续更高的温度(最终达110℃)进行真空干燥以除去残余的异丙醇。这得到143mg(69.7%收率)的白色固体,mp153.5-157℃。1HNMR(300MHz,D2O)δ8.29(s,1H),7.79(d,1H),7.63(d,1H),7.54(d,1H),7.34(m,2H),7.18(m,1H),7.10(s,1H),6.99(m,1H),4.05(d,1H),3.73(m,1H),3.44(m,2H),3.95-3.35(m,5H,包含樟脑磺酸),2.67(d,1H,樟脑磺酸),2.23(m,2H),1.70-2.10(m,8H,包含樟脑磺酸),1.46(m,1H,樟脑磺酸),1.25(m,1H,樟脑磺酸),0.84(s,3H,樟脑磺酸),0.64(s,3H,樟脑磺酸)。使用与上文所述的过程(实施例3-11)类似的过程,表征了几种其它的盐形式。这些制备物的结果在实施例12-14中记载。实施例12:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺硫酸盐的合成硫酸盐从乙酸异丙酯和水的混合物中沉淀析出。MP278℃。1HNMR(400MHz,DMSO-d6)δ9.28(宽的s,1H,酰胺),8.56(dd,1H),8.24(t,1H),7.77(d,1H),7.74(d,1H),7.60(s,1H),7.40(m,2H),7.35(s,1H),7.21(m,1H),4.21(m,1H),3.93(m,2H),3.10-3.60(m,5H),2.05(m,3H),1.92(m,1H),1.73(m,1H)。实施例13:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺酮戊二酸盐的合成α-酮戊二酸盐从乙酸异丙酯中沉淀析出。MP177℃。1HNMR(400MHz,DMSO-d6)δ8.64(s,1H,酰胺),8.50(d,1H),8.20(d,1H),7.74(d,1H),7.70(d,1H),7.60(m,1H),7.45(m,1H),7.32(m,2H),7.18(m,1H),4.10(m,1H),3.78(m,2H),3.00-3.45(m,5H),2.81(m,2H,酮戊二酸),2.41(m,2H,酮戊二酸),1.96(m,3H),1.83(m,1H),1.60(m,1H)。实施例14:(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺马尿酸盐的合成马尿酸盐从丙酮中沉淀析出(吸湿性太强,以致于不能获得熔点)。1HNMR(400MHz,DMSO-d6)δ8.79(s,1H,酰胺),8.56(d,1H),8.44(s,1H,马尿酸),8.29(m,1H),7.87(m,2H,马尿酸),7.76(d,1H),7.65(m,1H),7.54(m,1H),7.49(m,4H,包含马尿酸),7.34(m,2H),7.21(m,1H),3.91(m,1H),3.74(m,2H),3.00-3.50(m,5H),2.80(m,2H,马尿酸),1.79(m,2H),1.60(m,2H),1.30(m,1H)。实施例15:分离(2R,3R)-和(2S,3S)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺和转化成半乳糖二酸盐通过旋转蒸发,将从分离(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺对甲苯磺酸盐(实施例2)得到的上清液的样品浓缩,用10%的氢氧化钠水溶液调节到pH10并用二氯甲烷提取。蒸发二氯甲烷提取物,将残余物(1.8g)溶解在含有0.5%二正丁胺的绝对乙醇(55mL)中。以每份0.25mL将该溶液注射到25cm×2.1cmAD手性HPLC柱上,用60:40:0.2的己烷/乙醇/二正丁胺(流速=30mL/min)洗脱,在270nm监控。分离在~7.5分钟洗脱出的洗脱物和在~13.5分钟洗脱出的洗脱物,在蒸发溶剂后分别得到0.48g(98%手性纯度)和0.47g(99%手性纯度)的无色油状物。两个NMR谱相同。1HNMR(300MHz,CDCl3)δ8.49(s,1H),8.45(d,1H),7.74(d,1H),7.52(m,4H),7.35(t,1H),7.20(dd,1H),7.05(d,1H),4.55(dt,1H),3.43(m,1H),3.22(m,1H),2.90(m,5H),2.09(m,1H),1.88(m,4H)。将每个游离碱样品在无水乙醇(10mL)中的温热的溶液用一当量的半乳糖二酸处理。在搅拌下,将得到的混合物在75℃加热5分钟并冷却到周围环境温度。抽滤收集得到的固体并真空干燥,分别得到0.65g(87%收率)和0.62g(85%收率)的白色粒状固体(在各自的情况下,mp200-205℃)。1HNMR(300MHz,D2O)δ8.38(s,1H),8.28(d,1H),7.94(d,1H),7.70(d,1H),7.59(d,1H),7.48(t,1H),7.40(m,1H),7.32(m,2H),4.42(m,1H),4.21(s,2H),3.87(s,2H),3.68(m,1H),3.35(m,6H),2.25(m,2H),2.02(m,3H)。实施例16:(2R,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺对氯苯甲酸盐的合成将固体对氯苯甲酸(46.8mg,0.299mmol)一次性加入到得自实施例15的先前洗脱的N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基]苯并呋喃-2-甲酰胺异构体(108mg,0.299mmol)在丙酮(10mL)中的溶液中。将该混合物升温到接近回流,持续30分钟并冷却到周围环境温度。无沉淀物形成,因此将溶液浓缩到其原来体积的约20%(加热板),此时开始形成结晶。将混合物冷却并用异丙醇(2mL)稀释。通过在周围环境温度缓慢蒸发溶剂将该混合物浓缩,收集得到的固体并干燥。这产生145mg(94%收率)的浅黄色结晶,mp150-152℃。1HNMR(300MHz,CDCl3)δ8.49(s,1H),8.38(d,1H),7.93(d,2H,对氯苯甲酸),7.67(m,2H),7.57(d,1H),7.45(m,1H),7.36(d,2H,对氯苯甲酸),7.30(m,1H),7.27(s,1H),7.16(m,1H),7.00(d,1H,酰胺),6.90(宽的s,季铵),4.62(m,1H),3.85(dd,1H),3.36(m,1H),2.95-3.25(m,5H),2.16(s,1H),1.70-2.10(m,4H)。该样品的X射线结晶学分析揭示了其绝对立体化学是2R,3R(参见图11A和11B)。因此通过排除法,在实施例15中的后一种被洗脱的异构体具有2S,3S的绝对构型。实施例17:用于立体异构体分析的手性色谱方法获得用于将四个立体异构体彼此分离的手性色谱方法被证实非常具有挑战性。最初的企图(使用己烷/异丙醇/三乙胺流动相)导致峰重叠并且未达到最佳峰形。将异丙醇换成乙醇并将三乙胺换成二正丁胺,改善了分辨率和峰形并缩短了运行时间。该方法详细描述如下:分析柱:AD(250×4.6mm,5μm)流动相:60:40:0.2的己烷/乙醇/二正丁胺注射体积:10μl流速:1.0毫升/分钟温度:20℃检测:在270nm的UV总运行时间:~25分钟洗脱顺序(RT):2S,3R(5.3分钟);2R,3S(7.3分钟);2R,3R(8.3分钟);2S,3S(12.1分钟)立体异构体类似物的代表性色谱如图12所示。实施例18:XRPD对本说明书描述的几种盐样品进行XRPD分析。提供了盐酸盐(图13)和甲苯磺酸盐(图14)的衍射图样。X射线粉末衍射(XRPD)收集了一个或两个仪器的X射线粉末衍射图样。在SiemensD5000衍射仪上收集了一些图样,该SiemensD5000衍射仪使用CuKα辐射(40kV,40mA),θ-θ测角器,V20发散度和接收缝隙,石墨二级单色器和闪烁计数器。使用经证明的金刚砂标准品(NIST1976)进行仪器的性能检查。使用原样粉末在周围环境条件下将运行样品制成平板样品。将约35mg的样品轻轻地填塞进被切成经过研磨的零-背景(510)硅片的腔内。在分析期间将样品在其自身平面内旋转,以0.05°步长从2°到42°范围扫描,每步4秒,使用CuKα1在BrukerAXSC2GADDS衍射仪上收集一些X射线粉末衍射图样,该BrukerAXSC2GADDS衍射仪使用CuKα辐射(40kV,40mA),自动化XYZ平台,用于样品自动定位的激光视频显微镜和HiStar2-维区域检测器。X射线光学系统包括与0.3mm的针孔准直仪连接的单一的多层镜。光束发散度(即,样品上X射线束的有效尺寸)是约4mm。使用θ-θ连续扫描方式,采用20cm的样品-检测器距离,这得到3.2°-30.0°的有效2θ范围。典型地,样品将暴露于X射线束下,历时120秒。使用不经研磨的原样粉末在周围环境条件下将运行样品制成平板样品。将约1-2mg的样品轻轻地按压在硅片上以获得扁平表面。在非周围环境条件下将运行样品设置在具有导热化合物的硅片上。然后以大约10℃/分钟将样品加热到合适的温度并随后等温保持约5分钟,然后开始收集数据。差示扫描量热法(DSC)在装备有50位自动进样器的TA仪器Q1000上收集DSC数据。使用经证明的铟对仪器进行能量校准和温度校准。典型地,将在针孔式铝锅中的0.5-1.5mg的每个样品以10℃/分钟从25℃加热到175-200℃。在样品上方保持30mL/min的氮气吹扫。热解重量分析法(TGA)在装备有16位自动进样器的TA仪器Q500TGA上收集TGA数据。使用经证明的Alumel对仪器进行温度校准。典型地,将5-10mg的每个样品装载到预先称皮重的铂坩埚和铝DSC锅中,并以10℃/min从周围环境温度加热到350℃。在样品上方保持60mL/min的氮气吹扫。偏振光显微术(PLM)在具有用于捕捉图像的数字摄像机的LeicaLM/DM偏振光显微镜上研究样品。将少量的每个样品置于载玻片上,将其置于浸油中并用盖玻片覆盖,尽可能使单个颗粒充分地分离。使用合适的放大倍数和连接至λ伪色过滤器的部分偏振光来观察样品。热台显微术(HSM)使用与Mettler-ToledoMTFP82HT热台和用于捕捉图像的数字摄像机组合的LeicaLM/DM偏振光显微镜进行热台显微术。将少量的每个样品置于载玻片上,使得单个颗粒尽可能充分地分开。使用合适的放大倍数和连接至λ伪光过滤器的部分偏振光来观察样品,同时以典型的10℃/分钟从周围环境温度进行加热。重量蒸气吸附(GVS)在一个或两个仪器上测定吸附等温线。一些样品使用受VTIFlowSystem4软件控制的VTICorporationSGA-100水分吸附分析仪进行实验。借助于Polyscience恒温浴将样品温度保持在25℃。通过混合干和湿氮气流控制湿度。使用精确度为+/-0.0001g的CahnDigitalRecordingBalanceD-200监控作为%RH的函数的重量改变。典型地,在周围环境条件下将10-20mg的样品置于称皮重的天平盘中。将样品在50℃干燥1小时。在25℃,以5%RH为间隔,在5-95%RH范围内绘制标准品吸附等温线,并且类似地,在25℃下,以5%RH为间隔,在95-5%的RH范围内绘制解吸等温线。对于每个%RH数据点,样品平衡标准在5分钟或180分钟的最大平衡时间内包括0.0100wt%。使用受CFRSorp软件控制的HidenIGASorp水分吸附分析仪获得一些吸附等温线。通过Huber再循环式水浴锅将样品温度保持在25℃。通过将干和湿氮气流混合来控制湿度,使用的总流速为250mL/min。通过位于样品附近的经校准的VaisalaRH探针(动态范围0-95%RH)测量相对湿度。通过测微天平(精确度±0.001mg)不间断地监控样品的相对于RH%的重量改变(质量减轻)。典型地,在周围环境条件将10-20mg的样品置于称皮重的网格不锈钢吊篮中。在40%RH和25℃(典型的周围环境条件)下进行样品的装载和卸载。如下所述绘制水分吸附等温线(2次扫描得出1个完整循环)。在25℃下,以10%RH为间隔,在0-90%的RH范围内,绘制标准等温线。GVS通用法参数参数值吸附-扫描140-90解吸/吸附-扫描285-干,干-40间隔(%RH)10扫描数2流速(mL/min)250温度(℃)25稳定性(℃/min)0.05最小吸附时间(小时)1最大吸附时间(小时)4模式AF2精确度(%)98软件使用最小二乘方最小化过程以及质量减轻模型,来预测渐近值。测量的质量减轻值必须处在软件预测值的5%范围内,然后选择下一个RH%值。最小平衡时间被设为1小时,最大平衡时间被设为4小时。典型地,在完成等温线后回收样品并通过XRPD再次进行分析。通过KarlFischer法(KF)测量水在使用HydranalCoulomatAG试剂和氩吹扫的MettlerToledoDL39库仑计上测量每个样品的水含量。将称重的固体样品引入到容器内的铂TGA盘上,该盘连接至subaseal以避免水进入。每次滴定使用大约10mg的样品,并且进行一式两份的测定。通过HPLC测定热动力学水溶解度通过将足够的化合物悬浮在0.25mL的水中以获得最大的最终浓度为≥10mg/mL的无母体形式的化合物来测定水溶解度。将悬浮液在25℃平衡24小时,然后测量pH。然后将悬浮液过滤通过玻璃纤维C过滤器,进入96孔板。然后将滤液进行101倍稀释。相对于约0.1mg/mL的在DMSO中的标准溶液进行HPLC定量。注射不同体积的标准品、经过稀释的和未经稀释的样品溶液。使用峰面积计算溶解度,通过积分在与标准品注射中的主峰相同的保留时间发现的峰而测定所述峰面积。如果在过滤器板中存在足够的固体,则收集XRPD。用于热动力学水溶解度方法的通用法的详细描述通过HPLC测定化学纯度在装备有二极管阵列检测器并使用ChemStation软件v9的AgilentHP1100串联系统上进行纯度分析。使用下文详述的两个方法之一。方法1方法2离子色谱法在使用ICNet软件v2.3的Metrohm861AdvancedCompactIC上收集数据。将样品制备成在水中的1000ppm母液。当样品的溶解度低时,使用合适的共溶剂,诸如DMSO。在试验之前,使用合适的溶剂将样品稀释到50ppm或100ppm。通过与已知浓度的正被分析的离子的标准溶液相比来进行定量测定。阴离子的离子色谱法阳离子的离子色谱法将约50mg的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺盐酸盐在玻璃小瓶中称重并加热到50℃。向固体中加入100μl小份的1-丁醇/水(5体积%的水)直到形成澄清溶液(总共500μl)。将样品在50℃搅拌1小时并进行观测。在50℃加热1小时后,样品仍然是澄清溶液并以1.4℃/小时的速率将其从50℃冷却到25℃。样品在冷却时仍然是澄清溶液,并用带针孔的石蜡膜覆盖,在周围环境温度下允许其蒸发。在2周后,在经过部分蒸发的样品中可见大结晶。图13是(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐的XRPD,显示了观测图样(较亮)和计算图样(较暗)。实验图样得自(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺一盐酸盐的样品,而计算实例则得自本说明书所述的单晶X射线结构,并且描述于图10A和10B中。这两个图形在2θ值方面都是一致的,并且在强度和峰宽的微小差异可归因于仪器分辨率和优选取向的影响。另外,微小差异可归因于由于在室温下被收集的观察数据和收集在120K下的结构的计算数据所导致的温度变动。甲苯磺酸盐,特别是结晶单盐,经过证实并且使用CuKα辐射(40kV,40mA),θ-θ测角器,V20发散度和接收狭缝,石墨二级单色器和闪烁计数器获得的衍射图样如图14所示。在40℃/75%RH下历时1周后甲苯磺酸盐的XRPD衍射图揭示了改变,但是样品仍然是1型。这一改变很可能归因于更大程度的水合形式。VII.生物学试验化合物A及其立体异构体结合并调节各种NNR亚型的功能的能力根据Mazurov等人的美国专利6,953,855所述进行评价,通过引用将该文献的内容并入本说明书。化合物A的受体选择性表征(包括5HT3受体和毒蕈碱性受体)由BiosciencesCorporation进行。在以下的两个表达系统内进行α7NNR应答的电生理学测量:在哺乳动物GH4C1细胞中的大鼠α7NNR和在非洲爪蟾(Xenopus)卵母细胞中的人α7NNR。根据Placzek等人,Mol.Pharm.68(6):1863-1876(2005)所述,制备了表达大鼠α7NNR的GH4C1细胞,通过引用将该文献并入本说明书。使用流体动力快速灌注系统和膜片钳,使用该GH4C1细胞表达系统,实现了激动剂活性的电生理学测量。乙酰胆碱和烟碱都产生由α7介导的电流的浓度依赖性激活。得自文献的激动剂EC50值与使用该方法获得的那些EC50值是可比较的(参见Dunlop等人BiochemPharmacol付梓(2007)和Dynaflowonlinematerials(www.cellectricon.com),各自作为关于这一方法的参考被引用并入)。用Axopatch700A放大器记录的全细胞电流在1kHz下进行过滤并使用PCI卡片(NationalInstrument)在5kHz下取样。与先前的研究相比,根据所述来调节盐水溶液以增加电流稳定性。在室温下在以下的细胞外培养基中对细胞进行记录:130mMNaCl,5mMKCl,2mMCaCl2,2mMMgCl2,10mMHEPES,用含水NaOH调节到pH7.4。将硼硅酸盐电极(3-5MΩ)充满以下培养基:130mMTRIS磷酸盐,5mMNaCl,2mMMgCl2,10mMHEPES,10mMEGTA,用含水KOH调节到pH7.4(参见,Wu等人,J.Physiol.576:103-118(2006),该文献作于关于这一教导的参考被引用并入)。在这些条件下,当用1000μM乙酰胆碱(ACh)浓度引发时,使用NNR全细胞记录获得的大电流活性持续长达60分钟。采用得自Dynaflow的Cellectricon应用注释的细胞处理程序。简而言之,将细胞从温育箱中取出后,将细胞用记录介质充分洗涤三次并置于倒置Zeiss显微镜的平台上。在建立全细胞记录结构之前,平均5分钟是必要的。为了避免改变细胞条件,将每单一细胞负载的单一细胞记录进Dynaflow硅片中。在实验条件下可以检测到在应答性细胞的级分之间没有差异。大于95%的细胞对ACh应答,并且凡是提供可测量电流的细胞都被考虑在内。在整个实验中细胞被保持在-60mV。所有的供试制品溶液每天从储备溶液被制得。新鲜的乙酰胆碱(ACh)储备溶液每天在林格氏液中被制备并进行稀释。使用Prism5.0软件,通过单一Hill方程描绘剂量反应曲线。根据通过引用将其并入本说明书的Papke和Papke的Brit.J.Pharmacol.137:49-61(2002)所述,制备了表达人α7NNR的非洲爪蟾卵母细胞。使用成熟(>9cm)的雌性滑爪蟾(Xenopuslaevis)非洲蟾蜍(Nasco,Ft.Atkinson,WI)作为卵母细胞的来源。在手术前,通过将该动物置于1.5g/L的3-氨基苯甲酸乙酯的溶液中历时30分钟而将蟾蜍麻醉。从在腹部制造的切口中取出卵母细胞。为了除去卵泡细胞层,在室温下,在不含钙的Barth's溶液(88mMNaCl,1mMKCl,15mMHEPESpH7.6,0.81mMMgSO4,2.38mMNaHCO3,0.1mg/mL硫酸庆大霉素)中,将收获的卵母细胞用得自WorthingtonBiochemicalCorporation(Freehold,NJ)的1.25mg/mL胶原酶处理2小时。然后,分离出5期(stage)卵母细胞并用各自为50nL(5-20ng)的人α7cRNA注射该细胞。在注射后第2-7天进行记录。每天在林格氏液中制备新鲜的乙酰胆碱(ACh)储备溶液。使用OpusXpress6000A(AxonInstruments,UnionCityCA)进行实验。OpusXpress是提供最多八个并联的卵母细胞的自动化刺穿和电压钳的集成系统。电压和电流电极都充满3M的KCl。在-60mV的保持电位下对细胞进行电压钳试验。在50Hz下收集数据并在20Hz下进行过滤。将细胞用林格氏液进行浴灌注,并借助于一次性尖头从96孔板中释放激动剂溶液,这消除了任何交叉污染的可能性。流速被设定为2ml/min。药物施加在ACh对照和实验激动剂之间交替进行。施加是12秒的持续期、然后是181秒的间歇期。α7受体的应答被计算为净电荷(参见,如上所述的Papke和Papke,Brit.J.Pharmacol.137:49-61(2002))。每个卵母细胞接受最初的ACh的对照施加,然后接受实验药物施加,然后接受随后的ACh(300μM)对照施加。对实验药物施加的应答相对于上一次的ACh对照应答进行计算,以便对数据归一化,补偿在卵母细胞之间的变化的通道表达水平。注意,300μMACh激发得自α7受体的最大净电荷应答,以便向ACh对照的归一化有效地将数据归一化到ACh最大应答。从每个实验的浓度的至少四个卵母细胞的经归一化的应答计算平均数和标准误差(SEM)。对于浓度-应答关系,使用Kaleidagraph3.0.2(AbelbeckSoftware;Reading,PA)将从净电荷分析得到的数据进行绘图,并从Hill方程式产生曲线。根据以下规程进行化合物A的行为表征。根据通过引用将其并入本说明书的Ennaceur和Delacour的Behav.BrainRes.100:85-92(1988)的说明进行物体识别(OR)任务。根据通过引用将其并入本说明书的Levin等人的Behav.Pharm.10:675-680(1999)的说明进行放射状臂形迷宫(RAM)模型。根据Suemaru等人的Brit.J.Pharmacol.142(5):843-850(2004)的说明进行预脉冲抑制(PPI)试验。根据Roux等人的Curr.ProtocolsinPharmacol.Unit5.17(1999)的说明进行由阿扑吗啡诱导的运动活力(APOLOCO)的反转试验。体外生物活性总结化合物A以~1nM的平衡常数(Ki)竞争性抑制放射性标记的MLA与大鼠脑海马α7NNR的结合,显示了其对于α7NNR亚型具有非常高的亲合性。化合物A的立体异构体在大鼠α7NNR处具有以下的Ki值:2R,3S(42nM)[先前报道为28nM];2R,3R(1nM);2S,3S(25μM)(参见图1A)。如图1A2所示,化合物A的2S,3R对映体在α7亚型处显示与其它三种对映体类似物相反的活性,其它三种对映体类似物作为具有弱活性的重叠点被提供。化合物A不具有任何显著的亲合性(Ki值>2μM)地结合于α4β2NNR。使用在GH4C1(哺乳动物)细胞中稳定表达的大鼠α7NNR进行的膜片钳电生理学技术考察了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐(化合物A)及其立体异构体的功能活性。在这些实验中,化合物A与其它单独的异构体和所有四种异构体的外消旋混合物相比,产生了显著不同的功能性质。从图1A和1B可见,化合物A在引发功能应答方面(Emax=93%,相对于乙酰胆碱(ACh);EC50=14nM)比任一种其它的异构体或四种异构体的混合物具有更强的功效和更有效。实际上,化合物A(2S,3R异构体)是N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的唯一的能够在1-50nM的整个浓度范围内提供强效的激动作用的异构体,采用与本说明书所述的体内活性有关的10nM,如图1B所示。化合物A的功能活性还在瞬时表达人α7NNR的非洲爪蟾卵母细胞中进行了电生理学评价。在该系统中,化合物A具有33nM的EC50值和100%ACh应答的Emax。在施用浓度大于100nM(IC50=200nM)的(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺后的随后的对ACh的对照应答中有增加。与先前所述的α7完全激动剂(参见Astles等人,CurrentDrugTargetsCNSNeurologicalDisorders1(4):337-348(2002),其作为关于这一报道的参考被并入本说明书)相反,(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺的EC50和IC50之间的差别表明产生α7的半数最大功能应答的浓度导致最小的而非完全的残余抑制。当将(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺施用于表达人α4β2亚型的卵母细胞时没有可检测的激活,并且在随后的对ACh的对照应答中没有显著降低,表明(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在α4β2处既不是激动剂也不是拮抗剂。化合物在携带肌肉型受体(在人TE671/RD克隆细胞中的α1β1γδ亚型)、或神经节型受体(在大鼠嗜铬细胞瘤PC12细胞中和在人SHSY-5Y克隆细胞中的α3β4亚型)的功能模型中表现出很少的激动剂活性或不表现出激动剂活性,在这些亚型处产生≤10%(人肌肉),≤20%(大鼠神经节)和≤10%(人神经节)的烟碱应答。这些数据显示了对CNS亚型的选择性高于对PNS亚型的选择性。由于在α7和5-羟色胺(5HT3)受体之间的序列接近和结构同源性,以及使用其它的烟碱样配体观察到的对这两种受体的交叉反应性,考察了化合物A对5HT3受体的亲合性。化合物A(10μM)显示了在小鼠5HT3受体处的放射性配体结合的59%抑制和在人受体处的放射性配体结合的25%抑制。对在人5HT3受体处的功能激活的考察暗示了最小激活到无激活(即,在100μM得到15%激活的最大应答)。毒蕈碱性受体由于已经观察到的与其它烟碱样配体的相互作用而成为另一个有意义的领域。当在对M1、M2、非选择性中心毒蕈碱性受体和非选择性外周毒蕈碱性受体的竞争性结合抑制试验中进行考察时,化合物A显示最小的相互作用到无相互作用。数据显示化合物A对α7NNR配体具有选择性。化合物A在周围神经系统所特有的烟碱样受体的这些亚型处或在毒蕈碱性受体或5HT35-羟色胺能受体处不发生良好结合。因此,化合物A在治疗中枢神经系统障碍中具有治疗潜力,并且不产生与周围神经系统的相互作用有关的副作用。体内生物活性总结化合物A(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺或其药学上可接受的盐在两个认知行为模型中显示显著的功效。化合物A在大鼠的物体识别模型中,在i.p.(腹膜内给药,图3)和p.o.(口服给药,图4)后显示强效的活性,还在口服给药后的宽的剂量范围内显示活性(图4)。在相同的低剂量(0.3和1mg/kg)下腹膜内给药的化合物A倾向于反转在OR任务中由MK-801诱导的缺陷(图5),并且以0.3mg/kg被口服给药的化合物A具有持续至少18小时的认知作用(图6)。在考查工作记忆的放射状臂形迷宫(RAM)(图7)模型中,化合物A显著增加在错误选择之前的正确选择数。这些结果显示了(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺在治疗与精神分裂症有关的认知缺陷和功能障碍(包括与工作记忆有关的功能障碍)方面具有潜力。对于适用于治疗精神分裂症中的认知功能障碍的化合物,其不可削弱对抗精神分裂症的阳性症状的典型性或非典型性抗精神病药的影响。因此,除了化合物A的认知增强性质之外,令人注目的是,化合物A还有效地反转由阿扑吗啡诱导的运动活力(APOLOCO)(图8),并在精神分裂症的阳性症状的预脉冲抑制(PPI)(图9)模型中有效。因此,(2S,3R)-N-(2-((3-吡啶基)甲基)-1-氮杂双环[2.2.2]辛-3-基)苯并呋喃-2-甲酰胺将被预期在对抗与精神分裂症有关的阳性症状以及认知症状中提供额外的益处。观察到的特定的药理学应答可根据或取决于被选择的具体活性化合物,或者是否存在药学载体,以及所采用的制剂类型和给药方式而变化,并且根据本发明的实践,在结果中的这些被预期的变化或差异被本发明所涵盖。尽管已经说明和详细描述了本发明的具体的实施方案,但是本发明不限于这些具体的实施方案。上述的详细描说明作为本发明的示例而被提供,不应认为对本发明构成任何的限制。修改对本领域技术人员来说是明显的,并且这些修饰都不脱离本发明的精神,并且意在被所附权利要求书的范围所包括。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1