一类苯并噻唑衍生物及其制备方法与流程
本发明属于新材料技术领域,具体涉及一类苯并噻唑衍生物及其制备方法。
(二)
背景技术:
有机荧光材料被广泛应用于光学电子器件、DNA诊断、光化学传感器、染料、荧光增白剂、荧光涂料、激光染料、有机电致发光器件(OELD)等领域。与传统的无机材料相比,有机非线性光学材料具有优异的光学性能、原料获取广泛、制备低成本、良好的可加工性等无可比拟的优势,引起了很多人的关注,具有很大的潜在实用价值。
由于噻唑环S,N易配位的特性,噻唑环的衍生可用来制备离子探针、DNA探针,也可以用于分离元素rhenium(III),节省资源。苯并噻唑中硫原子易极化,使得分子轨道中电子易转移,所以在光化学上作用出色。
苯并噻唑类衍生物的合成硫原子引入是关键步骤,选择合适的反应原料对于苯并噻唑类衍生物的设计与合成来说至关重要。以邻氨基苯硫酚制备苯并噻唑类衍生物,但难以获得取代多样的邻氨基苯硫酚,随后又出现硫代酰胺或硫脲.由于邻氨基苯硫酚不稳定,很容易氧化,而硫代酰胺或硫脲这类原料的合成步骤比较长、制备成本比较高。
(三)、
技术实现要素:
本发明的第一个目的是提供一类苯并噻唑衍生物,是一类光学性能优异、稳定性好的有机荧光材料。本发明以苯并噻唑作为共轭单元,对其结构进行修饰得到了一类可在250-400nm范围内具特定吸收的苯并噻唑衍生物。Jin报道了用2-(2-羟基-苯基)苯并噻唑衍生物检测Zn2+,其stokes位移只有80nm,而本文所合成结构其stokes位移高达188nm。本发明采用的技术方案是:
本发明提供一类式(Ⅰ)所示苯并噻唑衍生物:
式(I)中:R1为氢、烷基或烷氧基;R2为氢或卤素原子。
作为优选,所述化合物为下列之一:
本发明的第二个目的是提供一种制备苯并噻唑衍生物(式Ⅰ所示化合物)的方法,所述制备方法包括如下步骤:
(1)将硫化试剂在溶剂A中回流1~2h后,将其加入到装有溶剂A和化合物(II)的反应容器中,回流搅拌2~4h,过滤得到黄色固体(III),反应式如下:
(2)将化合物(III)与溶剂B和还原剂在100℃~120℃反应5~7h,等还原剂消耗完毕加入肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为15:1石油醚和乙酸乙酯过柱得到式(IV)所示的2-苯乙烯基苯并噻唑衍生物,反应式如下:
其中R2为氢或卤素原子;
(3)取化合物(IV)加入到反应容器中,加入碱和化合物(IV)质量0.1倍的催化剂,然后再加入溶剂A,氩气氛围中回流24h后用体积比为10:1的石油醚和乙酸乙酯=过柱分离得到产物(V);所述碱为碳酸钠或碳酸钾;催化剂为四(三苯基膦)钯或二(三苯基膦)二氯化钯;反应式如下:
其中R1为氢、烷基或烷氧基;R2为氢或卤素原子。
作为优选,步骤(1)中所述的硫化试剂选自硫、硫化钠、硫化钾和二氧化硫中的两种;所述的溶剂A为甲醇、乙醇或丙醇,所述两种硫化试剂与化合物(II)的物质的量比为1:1:2~4,所述硫化试剂在溶剂A的质量比为1:2.52:95.8~100,所述溶剂A和化合物(II)的质量比为8.8~9:1。
作为优选,步骤(2)中所述溶剂B为醋酸、盐酸或硫酸;所述还原剂为锌粉或锡粉,化合物(III)、还原剂和肉桂醛衍生物的摩尔比为1:89.6:1~2,所述化合物(III)与溶剂B的质量比为1:525~530。
作为优选,步骤(3)中所述溶剂A为乙醇、甲醇或醇;所述化合物(IV)、碱和催化剂的物质的量比为1:8~10:0.1,所述溶剂A与化合物化合物(IV)的质量比为59~62:1。
本发明的第三个目的是提供一种苯并噻唑衍生物在荧光材料中的应用。本发明的目的是在苯并噻唑上引入溴原子,合成新型的苯并噻唑衍生物并应用到光学材料中。
本发明的有益效果:
本发明以苯并噻唑作为共轭单元,对其结构进行修饰,得到了一类在可250-400nm范围内具特定吸收的苯并噻唑衍生物,所得苯并噻唑衍生物光学性能优异、稳定性好,能应用于荧光材料领域。
(四)附图说明
图1是为实施例1-4中制备的苯并噻唑衍生物在二氯甲烷中的紫外-可见吸收光谱图,其中:横坐标表示的是波长,单位为nm,纵坐标表示的是吸收强度,单位为1。
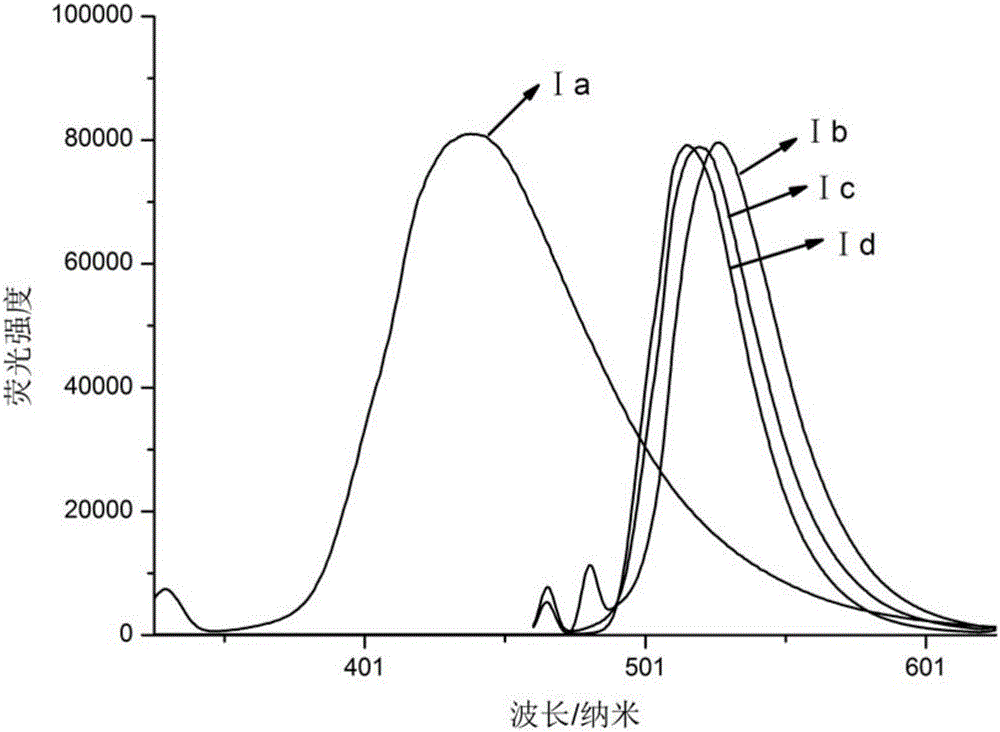
图2是为实施例1-4中制备的苯并噻唑衍生物在二氯甲烷中的荧光光谱图,其中:横坐标表示的是波长,单位为nm,纵坐标表示的是强度,单位为1。
(五)具体实施方式
下面结合具体实施例对本发明进行进一步描述,但本发明的保护范围并不仅限于此:
实施例1
(1)步骤一:
将4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h后,用恒压滴液漏斗加入到装有25ml甲醇和8mmol物质(II)的250ml圆底烧瓶中,回流搅拌4h,过滤得到黄色固体(III)(93mg),收率50%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml醋酸和1g锌粉后110℃反应6h后,等锌粉消耗完毕后加入11.33mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为50%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入8mmol的K2CO3和10%当量的Pd(PPh3)4,加入乙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到淡黄色固体产物(Va)2‐(4‐氟苯乙烯基)‐5‐((4‐甲氧基苯基)乙炔基)苯并噻唑,产率为60%。
m.p.183~185℃;1H NMR(500MHz,Chloroform-d)δ8.01(d,J=1.5Hz,1H),7.73(d,J=8.2Hz,1H),7.59–7.35(m,5H),7.35–7.15(m,1H),7.03(t,J=8.5Hz,2H),6.91–6.67(m,2H),3.76(s,3H).13C NMR(126MHz,CDCl3)δ167.56,159.55,153.51,136.70,133.84,132.90,131.31,131.28,129.08,129.01,128.33,125.23,121.65,121.34,121.25,115.96,115.78,114.88,113.87,89.64,87.52,77.28,77.22,77.03,76.77,55.11.HR-ESI/MS(m/z)calcd for C24 H17 F N O S[M+H]+=386.1009 found386.1022.
实施例2
(1)步骤一:
将4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h后,用恒压滴液漏斗加入到装有25ml甲醇和8mmol物质(II)的250ml圆底烧瓶中,回流搅拌4h,过滤得到黄色固体(III)(93mg),收率50%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml醋酸和1g锌粉后110℃反应6h后,等锌粉消耗完毕后加入11.33mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为50%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入8mmol的K2CO3和10%当量的Pd(PPh3)4,加入乙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到淡黄色固体产物(Vb)5-((4-甲氧基苯基)乙炔基)-2-苯乙烯基苯并噻唑,产率为60%。
m.p.170~172℃;1H NMR(500MHz,Chloroform-d)δ8.13(d,J=1.4Hz,1H),7.81(d,J=8.3Hz,1H),7.63–7.57(m,2H),7.57–7.50(m,4H),7.47–7.38(m,4H),7.28(s,0H),6.92(d,J=8.7Hz,2H),3.85(s,3H).13C NMR(126MHz,CDCl3)δ167.88,159.74,153.90,138.15,135.30,134.14,133.14,133.12,129.57,128.98,128.59,128.54,127.47,125.63,121.96,121.85,121.36,115.22,114.06,89.77,87.80,77.28,77.23,77.03,76.78,55.32.HR-ESI/MS(m/z)calcd for C24 H18 N O S[M+H]+=352.1154,found368.1094.
实施例3
(1)步骤一:
将4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h后,用恒压滴液漏斗加入到装有25ml甲醇和8mmol物质(II)的250ml圆底烧瓶中,回流搅拌4h,过滤得到黄色固体(III)(93mg),收率50%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml醋酸和1g锌粉后110℃反应6h后,等锌粉消耗完毕后加入11.33mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为50%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入8mmol的K2CO3和10%当量的Pd(PPh3)4,加入乙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到淡黄色固体产物(Vc)2-苯乙烯基-5-(对甲苯基乙基)苯并噻唑,产率为60%。
m.p.166~168℃;1H NMR(500MHz,Chloroform-d)δ8.13(dd,J=3.9,1.4Hz,1H),7.80(dd,J=16.4,8.3Hz,1H),7.62–7.58(m,2H),7.57–7.51(m,1H),7.51–7.45(m,2H),7.45–7.36(m,3H),7.36–7.22(m,1H),7.18(dd,J=8.2,2.8Hz,2H),2.39(d,J=2.0Hz,4H),13C NMR(126MHz,CDCl3)δ167.88,153.87,138.54,138.16,135.26,134.28,131.54,131.52,129.56,129.13,128.96,128.85,128.57,128.43,128.41,127.99,127.45,126.45,125.71,125.50,121.92,121.68,121.39,121.37,120.00,89.92,88.41,77.28,77.23,77.03,76.77,21.51.HR-ESI/MS(m/z)calcd for C24 H18 N S[M+H]+=352.1154,found352.1146.
实施例4
(1)步骤一:
将4mmol的Na2S和4mmol的S在15ml甲醇溶液中回流1h后,用恒压滴液漏斗加入到装有25ml甲醇和8mmol物质(II)的250ml圆底烧瓶中,回流搅拌4h,过滤得到黄色固体(III)(93mg),收率50%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml醋酸和1g锌粉后110℃反应6h后,等锌粉消耗完毕后加入12.88mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为50%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入8mmol的K2CO3和10%当量的Pd(PPh3)4,加入乙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到淡黄色固体产物(Vd)5-(苯基乙炔基)-2-苯乙烯基苯并噻唑,产率为57.4%。
m.p.160~163℃;1H NMR(500MHz,Chloroform-d)δ8.16(d,J=1.5Hz,1H),7.84(d,J=8.2Hz,1H),7.62(d,J=1.6Hz,1H),7.61(d,J=2.1Hz,2H),7.59(d,J=1.7Hz,1H),7.57(s,0H),7.56(d,J=1.5Hz,1H),7.54(d,J=1.6Hz,1H),7.46–7.43(m,2H),7.42(d,J=4.2Hz,1H),7.41(d,J=1.2Hz,0H),7.40–7.37(m,3H),7.28(s,1H).13C NMR(126MHz,CDCl3)δ167.96,153.90,138.24,135.28,134.50,131.68,129.60,128.99,128.60,128.39,127.48,125.85,123.13,121.93,121.49,121.43,89.73,89.08,77.28,77.02,76.77.HR-ESI/MS(m/z)calcd for C23 H16 N S[M+H]+=338.0988,found338.0998.
实施例5
(1)步骤一:
将4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h后,用恒压滴液漏斗加入到装有25ml乙醇和16mmol物质(II)的250ml圆底烧瓶中,回流搅
拌2h,过滤得到黄色固体(III),收率16.8%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml盐酸和1.8g锡粉,然后在130℃反应8h,等锡粉消耗完毕后加入22.66mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为15%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入10mmol的Na2CO3和10%当量的[(C6H5)3P]2PdCl,加入丙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到淡黄色固体产物(Ve)5-(苯基乙炔基)-2-苯乙烯基苯并噻唑,产率为11.4%。
实施例6
(1)步骤一:
将4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h后,用恒压滴液漏斗加入到装有25ml乙醇和16mmol物质(II)的250ml圆底烧瓶中,回流搅拌2h,过滤得到黄色固体(III),收率16.8%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml盐酸和1.8g锡粉,然后在130℃反应8h,等锡粉消耗完毕后加入22.66mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为15%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入10mmol的Na2CO3和10%当量的[(C6H5)3P]2PdCl,加入丙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到产物(Vf)苯并噻唑衍生物,产率为14.4%。
实施例7
(1)步骤一:
将4mmol的K2S和4mmol的SO2在15ml乙醇溶液中回流2h后,用恒压滴液漏斗加入到装有25ml乙醇和16mmol物质(II)的250ml圆底烧瓶中,回流搅拌2h,过滤得到黄色固体(III),收率16.8%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml盐酸和1.8g锡粉,然后在130℃反应8h,等锡粉消耗完毕后加入22.66mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(IV)(2-苯乙烯基苯并噻唑衍生物),收率为15%。
(3)步骤三:
取物质(IV)1mmol加入到50ml三口烧瓶中,加入10mmol的Na2CO3和10%当量的[(C6H5)3P]2PdCl,加入丙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到产物(Vg)苯并噻唑衍生物,产率为12.6%。
实施例8
(1)步骤一:
将4mmol的K2S和SO2在15ml乙醇溶液中回流2h后,用恒压滴液漏斗加入到装有25ml乙醇和16mmol物质(II)的250ml圆底烧瓶中,回流搅拌2h,过滤得到黄色固体(III),收率16.8%。
(2)步骤二:
将40mg的物质(III)加入到100ml圆底烧瓶中,加入20ml盐酸和1.8g锡粉,然后在130℃反应8h,等锡粉消耗完毕后加入36.22mg的肉桂醛衍生物反应4h,然后用乙酸乙酯萃取,用体积比为石油醚:乙酸乙酯=15:1过柱得到物质(VI)(2-苯乙烯基苯并噻唑衍生物),收率为13.4%。
(3)步骤三:
取物质(VI)1mmol加入到50ml三口烧瓶中,加入10mmol的Na2CO3和10%当量的[(C6H5)3P]2PdCl,加入丙醇25ml,氩气氛围中回流24h后用体积比为石油醚:乙酸乙酯=10:1过柱分离得到产物(Vh)苯并噻唑衍生物,产率为12.4%。
实施例9
苯并噻唑衍生物的性能检测
紫外-可见吸收光谱见图1:将化合物配成浓度为C=4×10-6mol/L的溶液,溶剂为二氯甲烷,使用的仪器是Shimadzu UV-1800分光光度计。从图1紫外吸收光谱中可以看出,4a-4d的紫外光谱均呈现两个明显的最大吸收峰,出现在290nm处的为π→π*电子跃迁产生的吸收峰;出现在310nm左右吸收峰则源于分子与CH2Cl2溶剂作用吸收。荧光关谱见图2:将化合物配成浓度为C=4×10-6mol/L的溶液,溶剂为二氯甲烷,使用的仪器是Shimadzu RF-6000PC光谱仪。从图2荧光发射图中看出Ⅰa分子为甲氧基与氟的衍生取代,相比Ⅰb,Ⅰc,Ⅰd发射峰520nm,发射光谱Ⅰa蓝移,最大发射峰在450左右。由于Ⅰa中F为吸电子基,电子云集中,导致共轭平面改变,发生蓝移。
表1苯并噻唑衍生物紫外吸收与荧光
λmax表示最大吸收波长,Eex表示激发波长,Em表示发射波长,Stokes shift表示斯托克斯位移。
由于Ia中F为吸电子基,电子云集中,导致共轭平面改变,发生蓝移,导致stokes位移小于Ib,Ic,Id。
以上仅列举了本发明的优选实施方案,本发明的保护范围并不限制于此,本领域技术人员在本发明权利要求范围内所作的任何改变均落入本发明保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!