琥珀酸多西拉敏晶型S及其制备方法与流程
本发明属医药领域,具体涉及一种乙醇类抗组胺药物琥珀酸多西拉敏的晶型S及其制备方法。
背景技术:
琥珀酸多西拉敏是一种乙醇类抗组胺药物,具备抗组胺、抗胆碱作用以及显著的镇静安眠效果,适用于包括过敏性皮肤病、枯草热、过敏性鼻炎、哮喘性支气管炎等在内的多种病症;由于其可通过抑制中枢神经系统来产生睡意,因此也被用作针对短期性失眠的治疗药物。
近年来,药物分子的多晶型现象引起了科学家们的广泛关注,由于不同的多晶型态在稳定性、解离度、生物利用度等方面均可能有很大的差别,因此多晶型的研究越来越有必要。科研工作者在药物晶型领域不断探索,试图获得高稳定性、高解离度、高生物利用度的晶形,进而提高药物的生物活性。
目前报道的琥珀酸多西拉敏的晶型主要包括晶型I(琥珀酸多西拉敏1:1)、晶型II和晶型S(琥珀酸多西拉敏1.5:1),其中晶型II只在热显微镜载片上熔融后制得,晶型I则为稳定晶型,晶型S为不稳定晶型。但是,对晶型S的相关研究有助于在琥珀酸多西拉敏工业化生产过程中最大程度上避免转晶的发生。
现有文献中公开的晶型S的制备方法大多采用琥珀酸多西拉敏的有机溶剂进行析晶,容易形成混晶,导致晶型S的收率较低纯度较差。另外文献中公开的为重结晶的方法,收率会受到影响,且不易大规模制备。本发明的目的之一在于公开了一种易于制备、纯度高、重现性好的琥珀酸多西拉敏晶型S及其制备方法。
技术实现要素:
本发明公开了一种纯度高、重现性好的式(I)琥珀酸多西拉敏晶型S结晶形态,以下称为晶型S,还公开了琥珀酸多西拉敏晶型S的制备方法。
本发明所述的琥珀酸多西拉敏晶型S的X射线粉末衍射图在10.9°(±0.2°)、11.4°(±0.2°)、12.1°(±0.2°)、13.2°(±0.2°)、13.9°(±0.2°)、14.4°(±0.2°)、15.6°(±0.2°)、17.6°(±0.2°)、19.2°(±0.2°)、20.8°(±0.2°)、21.5°(±0.2°)、22.0°(±0.2°)、22.7°(±0.2°)、22.9°(±0.2°)、23.3°(±0.2°)、23.5°(±0.2°)、23.8°(±0.2°)和24.4°(±0.2°)2θ角有特征峰。
进一步的,使用Cu-Kα辐射检测的X射线粉末衍射图谱中,具有以下特征峰,其2θ角度值及相对强度如下表所示:
本发明所述的琥珀酸多西拉敏晶型S具有如附图1所示的X粉末衍射图谱。
本发明所述的琥珀酸多西拉敏晶型S,如图3所示,差示扫描量热法分析图谱显示在约96.3~99.1℃处有吸热峰,表明其在约96.3℃左右开始熔融,出现吸热峰。
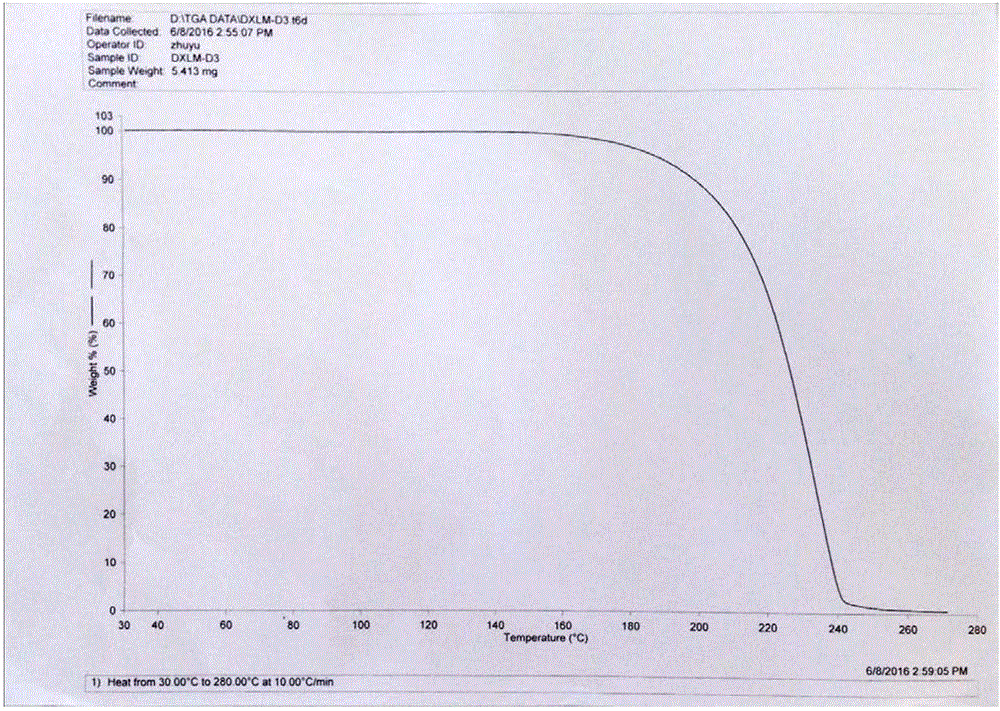
本发明所述的琥珀酸多西拉敏晶型S具有如图2、图3所示的热重分析图谱和差示扫描量热法分析图谱。
本发明同时提供制备琥珀酸多西拉敏晶型S的方法,该方法包括:
(a)将多西拉敏加热溶于一种有机溶剂中,并加入琥珀酸成盐;所述琥珀酸与多西拉
敏的配比为(1.1~1.7):1;
(b)缓慢降至0~25℃,析出固体,过滤干燥得到琥珀酸多西拉敏新晶型S。
本发明所述以上制备方法,步骤(a)中所述琥珀酸与多西拉敏的配比优选(1.4~1.6):1。
本发明所述以上制备方法,步骤(a)中使用的溶剂量与多西拉敏体质比(体积质量比)为(1.5-20):1,优选(2~8):1。
本发明所述以上制备方法,步骤(a)中使用的溶剂中含水量对晶型S的形成具有诱导作用,含水量范围为1.0%~5%,优选1.5%~3%。
本发明所述以上制备方法,步骤(a)中所述有机溶剂选自甲醇、乙醇、四氢呋喃、丙酮或乙酸乙酯,优选丙酮。
本发明所述制备方法,步骤(a)加热温度为40~70℃,步骤(b)析晶温度为0-25℃。
下表1为参照实施例1方法,采用丙酮作为溶剂变化物料配比和含水量,得到产品的情况。
表1配比及含水量对晶型的影响
备注:a:质量收率:实际获得产品质量占其加入反应器多西拉敏质量的百分数。
试验1-8数据显示:在溶剂无水情况下,当配比为1:0.95时体系倾向于形成晶型I,当配比为1:1.0~1:1.05时体系易形成混晶,当配比为1:1.0~1:1.5时体系倾向于形成晶型S,且产品收率逐渐增大,说明晶型S的形成主要受配比控制;但当琥珀酸配比进一步增大时,琥珀酸过量,易与产品形成混合物析出,影响产品质量。综上琥珀酸与游离碱的配比应控制在1:1.1~1.7,优选1:1.4~1.6。
对比试验1-3及试验9-11数据可以看出:含水量增大至2.5%时,当配比为1:0.95~1:1.05时体系则倾向于形成晶型S,且随着配比增大,产品收率均较不加水时较高,说明适量水分对晶型S的形成有诱导作用。
对比试验4-6及试验12-18数据可以看出,含水量增大时,相同投料比条件下,产品收率减小,这主要与产品在水中有较好的溶解性有关。因此含水量应控制在1.5%~5%。
有益效果
本发明提供的制备方法,能够高产率、高纯度制备得到琥珀酸多西拉敏晶型S产品。同时发现适量水分对晶型S的形成有诱导作用,能够提高产率。该方法操作简单、重现性好。
附图说明
图1为琥珀酸多西拉敏晶型S的X粉末衍射图谱;
图2为琥珀酸多西拉敏晶型S的热重分析图谱;
图3为琥珀酸多西拉敏晶型S的差示扫描量热法分析图谱。
具体实施方式
以下具体实施方式可以对本发明的内容做出更为详细的说明,但本发明的保护范围不局限于以下具体实施例。凡是基于本发明内容所实现的技术、工艺均属于本发明的范围。
一、琥珀酸多西拉敏晶型S制备方法如下反应方程式所示:
实施例1:
将20.0g的N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺于40~50℃溶于120mL丙酮(体质比1:6),搅拌加入13.1g琥珀酸,升温至60℃,搅拌反应0.5-1小时,缓慢降温至0-5℃,搅拌析晶3~5小时,在氮气保护下抽滤,滤饼用60mL 0~5℃丙酮洗涤,滤饼在≤-0.08Mp、45-55℃减压干燥3~4小时,得到N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺琥珀酸盐(1:1.5)类白色固体29.1g,收率87.9%,HPLC纯度99.8%。实施例2:
将20.0g的N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺于40~50℃溶于120mL丙酮(含5%的水)(体质比1:6),搅拌加入13.1g琥珀酸,升温至60℃,搅拌反应0.5-1小时,旋蒸至糊状,得橙黄色粘稠物,补加丙酮60mL,搅拌得类白色固体析出,在氮气保护下抽滤,滤饼用60mL 0~5℃丙酮洗涤,滤饼在≤-0.08Mp、45-55℃减压干燥3~4小时,得到N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺琥珀酸盐(1:1.5)类白色固体29.91g,收率90.3%,HPLC纯度99.8%。
上述方法制备得到的琥珀酸多西拉敏晶型S,X粉末衍射图谱如图1所示,热重分析图谱如图2所示,差示扫描量热法分析图谱如图3所示;
实施例3:
将20.0g的N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺于50~60℃溶于400mL乙酸乙酯(体质比1:20),搅拌加入13.1g琥珀酸,升温至70℃,搅拌反应0.5-1小时,缓慢降温至0-5℃,搅拌析晶2~3小时,在氮气保护下抽滤,滤饼用100mL 0~5℃乙酸乙酯洗涤,滤饼在≤-0.08Mp、45-55℃减压干燥3~4小时,得到N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺琥珀酸盐(1:1.5)类白色固体23.6g,收率17.3%,HPLC纯度99.6%。
实施例4:
将20.0g的N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺于50~60℃溶于60mL四氢呋喃(体质比1:3),搅拌加入13.1g琥珀酸,升温至66℃,搅拌反应0.5-1小时,缓慢降温至0-5℃,搅拌析晶2~3小时,在氮气保护下抽滤,滤饼用10mL 0~5℃四氢呋喃洗涤,滤饼在≤-0.08Mp、45-55℃减压干燥3~4小时,得到N,N-二甲基-2-[1-苯基-1-(2-吡啶)乙氧基]乙胺琥珀酸盐(1:1.5)类白色固体20.8g,收率62.8%,HPLC纯度99.8%。
- 还没有人留言评论。精彩留言会获得点赞!